You might also like
- Taxonomy Matching Using Background Knowledge: Linked Data, Semantic Web and Heterogeneous RepositoriesFrom EverandTaxonomy Matching Using Background Knowledge: Linked Data, Semantic Web and Heterogeneous RepositoriesNo ratings yet
- Data Mining Algorithms in C++: Data Patterns and Algorithms for Modern ApplicationsFrom EverandData Mining Algorithms in C++: Data Patterns and Algorithms for Modern ApplicationsNo ratings yet
- Project 5467Document13 pagesProject 5467stephennrobertNo ratings yet
- Multilevel Techniques For The Clustering ProblemDocument15 pagesMultilevel Techniques For The Clustering ProblemCS & ITNo ratings yet
- Brazilian Conformity Assessment System Intervention AnalyzedDocument26 pagesBrazilian Conformity Assessment System Intervention AnalyzedMarcos Pereira Estellita LinsNo ratings yet
- Computational Intelligence Based Approaches in Decision-MakingDocument5 pagesComputational Intelligence Based Approaches in Decision-MakingAlice DalinaNo ratings yet
- Fuzzy Coco: A Cooperative-Coevolutionary Approach To Fuzzy ModelingDocument11 pagesFuzzy Coco: A Cooperative-Coevolutionary Approach To Fuzzy ModelingIvonne Adriana ZuñigaNo ratings yet
- Paper - Hierarchical ClusterDocument13 pagesPaper - Hierarchical ClusterMuthia NadhiraNo ratings yet
- Ontology Matching Using An Artificial Neural Network To Learn WeightsDocument6 pagesOntology Matching Using An Artificial Neural Network To Learn WeightsDhanik ListianiNo ratings yet
- Hybrid Clustering Maximizes Gene SimilarityDocument14 pagesHybrid Clustering Maximizes Gene Similarityvmurali.infoNo ratings yet
- Casey Kevin MSThesisDocument51 pagesCasey Kevin MSThesisCourtney WilliamsNo ratings yet
- Ontology EnrichmentDocument10 pagesOntology EnrichmentRihab IdoudiNo ratings yet
- Clustering of Psychological Personality Tests of Criminal OffendersDocument8 pagesClustering of Psychological Personality Tests of Criminal OffendersakhtarrasoolNo ratings yet
- Bazan 2013Document44 pagesBazan 2013Dadang Syarif SSNo ratings yet
- Systems Biology and AutomataDocument9 pagesSystems Biology and AutomataCarlos CastañedaNo ratings yet
- NRMCB Review Accepted forRPSDocument41 pagesNRMCB Review Accepted forRPSMr. EmixiNo ratings yet
- Genetic Algorithm: Review and Application: Manoj Kumar, Mohammad Husian, Naveen Upreti & Deepti GuptaDocument4 pagesGenetic Algorithm: Review and Application: Manoj Kumar, Mohammad Husian, Naveen Upreti & Deepti GuptanelsonneymanNo ratings yet
- Evolutionary Computing Strategies For Gene SelectionDocument6 pagesEvolutionary Computing Strategies For Gene SelectionseventhsensegroupNo ratings yet
- Ant Colony Optimisation, Evolutionary Computation Including Genetic Algorithm, Iterated Local Search, Simulated Annealing and Tabu SearchDocument10 pagesAnt Colony Optimisation, Evolutionary Computation Including Genetic Algorithm, Iterated Local Search, Simulated Annealing and Tabu SearchAbidin ZeinNo ratings yet
- Discuss The Relationship Between Theory and Anlysis Through A Sample of Data From Your StudyDocument5 pagesDiscuss The Relationship Between Theory and Anlysis Through A Sample of Data From Your StudyInga Budadoy Naudadong100% (1)
- Multi-Objective Evolutionary Biclustering of Gene Expression DataDocument14 pagesMulti-Objective Evolutionary Biclustering of Gene Expression DatasfarithaNo ratings yet
- CP in Bioinformatics - 2019 - BeerliDocument28 pagesCP in Bioinformatics - 2019 - BeerliMaryam AsimNo ratings yet
- PROMPT: Algorithm and Tool For Automated Ontology Merging and AlignmentDocument6 pagesPROMPT: Algorithm and Tool For Automated Ontology Merging and AlignmentsamanNo ratings yet
- Biocluster MB05Document26 pagesBiocluster MB05DerrazNo ratings yet
- Multiple Clustering Views For Data AnalysisDocument4 pagesMultiple Clustering Views For Data AnalysisInternational Journal of Application or Innovation in Engineering & ManagementNo ratings yet
- Making Decision and Computer ScienceDocument10 pagesMaking Decision and Computer ScienceTrong-Minh HoangNo ratings yet
- Balancing Security Overhead and Performance Metrics Using A Novel Multi-Objective Genetic ApproachDocument7 pagesBalancing Security Overhead and Performance Metrics Using A Novel Multi-Objective Genetic ApproachInternational Journal of Application or Innovation in Engineering & ManagementNo ratings yet
- The Cooperative Royal Road: Avoiding HitchhikingDocument12 pagesThe Cooperative Royal Road: Avoiding HitchhikingMarius_2010No ratings yet
- Review of Multi-Omics Integration in The Age of Million Single-Cell DataDocument2 pagesReview of Multi-Omics Integration in The Age of Million Single-Cell Datazzh991209No ratings yet
- Challenges and Opportunities in Coding The Commons: Problems, Procedures, and Potential Solutions in Large-N Comparative Case StudiesDocument27 pagesChallenges and Opportunities in Coding The Commons: Problems, Procedures, and Potential Solutions in Large-N Comparative Case StudiesAlan HartogNo ratings yet
- Neural NeworkDocument23 pagesNeural NeworkJuan Peña ReyesNo ratings yet
- Strategies for Dealing with Real World Classification ProblemsDocument6 pagesStrategies for Dealing with Real World Classification ProblemsqazNo ratings yet
- An Analysis of Dematel Approaches For CriteriaDocument21 pagesAn Analysis of Dematel Approaches For CriteriaLeonel Gonçalves DuqueNo ratings yet
- Machine learning methods for survey researchersDocument10 pagesMachine learning methods for survey researchersRegistro Información Copia BackNo ratings yet
- Medical Data Mining Using Evolutionary ComputationDocument24 pagesMedical Data Mining Using Evolutionary ComputationAnonymous TxPyX8cNo ratings yet
- The Role of Text Pre-Processing in Sentiment Analysis: Information Technology and Quantitative Management (ITQM2013)Document7 pagesThe Role of Text Pre-Processing in Sentiment Analysis: Information Technology and Quantitative Management (ITQM2013)vishal3193No ratings yet
- The Semantic Similarity EnsembleDocument17 pagesThe Semantic Similarity EnsembleAndrea BallatoreNo ratings yet
- Collaborative Filtering Using RegressionDocument22 pagesCollaborative Filtering Using RegressionmuhammadrizNo ratings yet
- Logico-Ontological Framework Validates Research MethodsDocument17 pagesLogico-Ontological Framework Validates Research Methodsyashashree barhateNo ratings yet
- Discovering and Reconciling Value Conflicts For Data IntegrationDocument24 pagesDiscovering and Reconciling Value Conflicts For Data IntegrationPrakash SahuNo ratings yet
- Literature Mining in Molecular BiologyDocument5 pagesLiterature Mining in Molecular BiologyVishwadeep SinghNo ratings yet
- Mechanism Theory - Mat JacksonDocument46 pagesMechanism Theory - Mat JacksonAditya AryaNo ratings yet
- Analysis of Gene Microarray Data Using Association Rule MiningDocument5 pagesAnalysis of Gene Microarray Data Using Association Rule MiningJournal of ComputingNo ratings yet
- A Review of Feature Selection Methods On Synthetic DataDocument37 pagesA Review of Feature Selection Methods On Synthetic DataAllan HeckNo ratings yet
- Multivariate Exploratory Data AnalysisDocument2 pagesMultivariate Exploratory Data AnalysisMechanical Maths TutionNo ratings yet
- Ontology-Based Integration of InformationDocument10 pagesOntology-Based Integration of InformationHồ Văn BảoNo ratings yet
- 4968-Article Text-18207-1-10-20211015Document15 pages4968-Article Text-18207-1-10-20211015Aishwarya ANo ratings yet
- A Comparative Study On Mushrooms ClassificationDocument8 pagesA Comparative Study On Mushrooms ClassificationMuhammad Azhar Fairuzz HilohNo ratings yet
- Métodos de Integración de Datos para Descubrir Interacciones Genotipo-FenotipoDocument14 pagesMétodos de Integración de Datos para Descubrir Interacciones Genotipo-FenotipoKatherine ObeidNo ratings yet
- Co-Clustering Interpretations For Feature Selection by Using Sparsity LearningDocument6 pagesCo-Clustering Interpretations For Feature Selection by Using Sparsity LearningKumara SNo ratings yet
- Obust Model For Gene Anlysis and Classification: Fatemeh Aminzadeh, Bita Shadgar, Alireza OsarehDocument10 pagesObust Model For Gene Anlysis and Classification: Fatemeh Aminzadeh, Bita Shadgar, Alireza OsarehIJMAJournalNo ratings yet
- Uncertainty Analysis Using Fuzzy Sets For Decision Support SystemDocument19 pagesUncertainty Analysis Using Fuzzy Sets For Decision Support Systemaaaaa2012No ratings yet
- Contemporary Approaches To Mixed Methods-Grounded Theory Research: A Field-Based AnalysisDocument17 pagesContemporary Approaches To Mixed Methods-Grounded Theory Research: A Field-Based AnalysisALBA AZA HERNÁNDEZNo ratings yet
- Arbitrating Among Competing Classiers Using Learned RefereesDocument24 pagesArbitrating Among Competing Classiers Using Learned RefereesargamonNo ratings yet
- Expertise in Qualitative Prediction of Behaviour: Ph.D. Thesis (Chapter 0)Document21 pagesExpertise in Qualitative Prediction of Behaviour: Ph.D. Thesis (Chapter 0)Dart.srkNo ratings yet
- Coupling Simulation and Experiment - SHPSpartC - 2013Document31 pagesCoupling Simulation and Experiment - SHPSpartC - 2013Sameera HameedNo ratings yet
- An Evidence Based Approach To Collaborative Ontology DevelopmentDocument4 pagesAn Evidence Based Approach To Collaborative Ontology DevelopmentproconteNo ratings yet
- Scinem: A Scalable Data Science Tool For Heterogeneous Network MiningDocument4 pagesScinem: A Scalable Data Science Tool For Heterogeneous Network MiningLab George BrownNo ratings yet
- Articulo 8 PDFDocument14 pagesArticulo 8 PDFFabio Andres AgudeloNo ratings yet
- Articulo 9 PDFDocument6 pagesArticulo 9 PDFFabio Andres AgudeloNo ratings yet
- Articulo 5 PDFDocument13 pagesArticulo 5 PDFFabio Andres AgudeloNo ratings yet
- Articulo 7 PDFDocument22 pagesArticulo 7 PDFFabio Andres AgudeloNo ratings yet
- Articulo 6 PDFDocument9 pagesArticulo 6 PDFFabio Andres AgudeloNo ratings yet
- Metaheuristics Overview Blum PDFDocument41 pagesMetaheuristics Overview Blum PDFSamuel VictorinoNo ratings yet
- MRT Laboratory: Operation Manual For MRT Data ExplorerDocument105 pagesMRT Laboratory: Operation Manual For MRT Data Exploreralbaneza2 albanesNo ratings yet
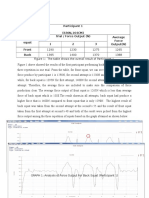
- Result: Participant 1 (630N, 164CM) Type of Squat Trial / Force Output (N) Average Force Output (N) 1 2 3 Front BackDocument5 pagesResult: Participant 1 (630N, 164CM) Type of Squat Trial / Force Output (N) Average Force Output (N) 1 2 3 Front BackpatenggNo ratings yet
- BBS Model Question 2020Document44 pagesBBS Model Question 2020JALDIMAINo ratings yet
- Thermal PhysicsDocument29 pagesThermal PhysicsAnonymous rn5Te9MwkNo ratings yet
- User's Guide Materials Control: ProductionDocument52 pagesUser's Guide Materials Control: ProductionRanko LazeskiNo ratings yet
- Inferences About Process QualityDocument108 pagesInferences About Process QualityguaritooNo ratings yet
- MARE3Document42 pagesMARE3chuchu maneNo ratings yet
- Solved Probability ProblemsDocument7 pagesSolved Probability ProblemsTHEOPHILUS ATO FLETCHERNo ratings yet
- Strcteng200 S1 2021Document11 pagesStrcteng200 S1 2021ChengNo ratings yet
- Drawing SheetsDocument11 pagesDrawing SheetsMona DiaaNo ratings yet
- Training Feed Forward Networks With The Marquardt AlgorithmDocument5 pagesTraining Feed Forward Networks With The Marquardt AlgorithmsamijabaNo ratings yet
- Lilo-An National High SchoolDocument3 pagesLilo-An National High SchoolJose Benavente100% (1)
- Methods of Analysis and Solutions of Crack ProblemsDocument562 pagesMethods of Analysis and Solutions of Crack ProblemsgedysonlimaNo ratings yet
- Shree MetriDocument18 pagesShree Metrivireshsa789No ratings yet
- MATH 4 SLM Week 2Document10 pagesMATH 4 SLM Week 2Dan August A. GalliguezNo ratings yet
- Structural calculations for holding tank shoring worksDocument9 pagesStructural calculations for holding tank shoring worksJohn BuntalesNo ratings yet
- MWD Log Quality & Standards - BHI - 1996 PDFDocument168 pagesMWD Log Quality & Standards - BHI - 1996 PDFsamanNo ratings yet
- Charly ChalkboardDocument9 pagesCharly ChalkboardOscar PachecoNo ratings yet
- Logic 3Document33 pagesLogic 3Kim Abigelle GarciaNo ratings yet
- Combo FixDocument4 pagesCombo FixJulio Cesar Camones CastilloNo ratings yet
- ECE 2303 Soil Mech 1 GR A Batch 1 PDFDocument32 pagesECE 2303 Soil Mech 1 GR A Batch 1 PDFCalebNo ratings yet
- Chap 4Document17 pagesChap 4Yuanjing XuNo ratings yet
- Powder FlowDocument15 pagesPowder FlowSteven SastradiNo ratings yet
- g9 Tle q3 Module 8 9 Week 5 6 Tech DraftDocument18 pagesg9 Tle q3 Module 8 9 Week 5 6 Tech Draftesller50% (2)
- Credit Card Fraud Detection Using Adaboost and Majority VotingDocument4 pagesCredit Card Fraud Detection Using Adaboost and Majority VotingVasavi DaramNo ratings yet
- JVC RX Es1slDocument20 pagesJVC RX Es1slvideosonNo ratings yet
- Checklist Design ReviewDocument2 pagesChecklist Design Reviewmuhannad11061975100% (1)
- Preprint Xyz IsmcDocument9 pagesPreprint Xyz IsmcThien MaiNo ratings yet
- PEQ - Acids & Alkalis - KS3 (PH)Document3 pagesPEQ - Acids & Alkalis - KS3 (PH)yayaNo ratings yet