You might also like
- Precipitation Hardening: The Commonwealth and International Library: Selected Readings in MetallurgyFrom EverandPrecipitation Hardening: The Commonwealth and International Library: Selected Readings in MetallurgyNo ratings yet
- Corrosion-Resistant Component For PEM Fuel Cells: Shuo-Jen Lee, Ching-Han Huang, Jian-Jang Lai, Yu-Pang ChenDocument7 pagesCorrosion-Resistant Component For PEM Fuel Cells: Shuo-Jen Lee, Ching-Han Huang, Jian-Jang Lai, Yu-Pang ChenSoh Ming LunNo ratings yet
- Materials Chemistry and PhysicsDocument11 pagesMaterials Chemistry and PhysicsAbraham Becerra AranedaNo ratings yet
- Power PlantDocument17 pagesPower PlantVasu RajaNo ratings yet
- Thin Solid Films, 517 5 (2009) 1672-1676Document5 pagesThin Solid Films, 517 5 (2009) 1672-1676Krishnan RamaswamyNo ratings yet
- Corrosion Science 50 (2008) 2620–2634Document15 pagesCorrosion Science 50 (2008) 2620–2634pepe martinezNo ratings yet
- Materials and Design: Haixu Li, Hao Yu, Tao Zhou, Baoliang Yin, Shaojiang Yin, Yanling ZhangDocument9 pagesMaterials and Design: Haixu Li, Hao Yu, Tao Zhou, Baoliang Yin, Shaojiang Yin, Yanling ZhangJose David CastroNo ratings yet
- Characterization of Native and Anodic Ox PDFDocument7 pagesCharacterization of Native and Anodic Ox PDFEdinei PaivaNo ratings yet
- Corrosion Resistance of Si-Al-bearing Ultrafine-Grained Weathering SteelDocument8 pagesCorrosion Resistance of Si-Al-bearing Ultrafine-Grained Weathering SteelGanesh.RajanNo ratings yet
- Pan Ago Poulos 2009Document6 pagesPan Ago Poulos 2009HARI KRISHNAN GNo ratings yet
- 1-s2.0-S0022311518300588-mainDocument8 pages1-s2.0-S0022311518300588-mainpela210No ratings yet
- Effect of Microcrystallization On Pitting Corrosion of Pure AluminiumDocument7 pagesEffect of Microcrystallization On Pitting Corrosion of Pure Aluminiumh.mraiedNo ratings yet
- tang2012Document12 pagestang2012bokeya7674No ratings yet
- Self-Ion Bombarded CR Films Crystallographic Orientation and Oxidation BehaviourDocument9 pagesSelf-Ion Bombarded CR Films Crystallographic Orientation and Oxidation BehaviourBeeNo ratings yet
- Glass Coatings Protect Stainless Steel from High-Temperature OxidationDocument12 pagesGlass Coatings Protect Stainless Steel from High-Temperature OxidationJabbar AljanabyNo ratings yet
- CIMNAS Article 14 - Preprint VersionDocument16 pagesCIMNAS Article 14 - Preprint VersionAqeel razaqNo ratings yet
- cr500002z PDFDocument70 pagescr500002z PDFElisa FoundaNo ratings yet
- Corrosion Science: M. Buc Ko, J. Rogan, S.I. Stevanovic, A. Peric - Grujic, J.B. BajatDocument11 pagesCorrosion Science: M. Buc Ko, J. Rogan, S.I. Stevanovic, A. Peric - Grujic, J.B. BajatterNo ratings yet
- Effect of Heat Treatment and Bath Composition of Electroless Nickel-Plating On Cavitation Erosion ResistanceDocument23 pagesEffect of Heat Treatment and Bath Composition of Electroless Nickel-Plating On Cavitation Erosion ResistanceSantiago TuestaNo ratings yet
- Corrosion Science: Rajiv P. Edavan, Richard KopinskiDocument14 pagesCorrosion Science: Rajiv P. Edavan, Richard KopinskiVanessa Rios NolayaNo ratings yet
- Pitting Corrosion of AluminumDocument25 pagesPitting Corrosion of AluminumBeatriz BrachettiNo ratings yet
- native oxide for XPSDocument12 pagesnative oxide for XPSParth KhandelwalNo ratings yet
- The Effects of Film Thickness and Incorporated Anions On Pitting Corrosion of Aluminum With Barrier-Type Oxide Films Formed in Neutral Borate and Phosphate ElectrolytesDocument8 pagesThe Effects of Film Thickness and Incorporated Anions On Pitting Corrosion of Aluminum With Barrier-Type Oxide Films Formed in Neutral Borate and Phosphate ElectrolytesDamon CiouNo ratings yet
- Surface & Coatings Technology: Hung-Bin Lee, Tzu-Jing Lin, Chun-Ying LeeDocument12 pagesSurface & Coatings Technology: Hung-Bin Lee, Tzu-Jing Lin, Chun-Ying LeeFrancisco OppsNo ratings yet
- Electrochemical Behavior and Passive Film Properties of Co-Cr-Mo Stainless SteelDocument7 pagesElectrochemical Behavior and Passive Film Properties of Co-Cr-Mo Stainless SteelEr Dikshant MalhotraNo ratings yet
- Surface Characterisation and Crevice Corrosion Behaviour of Nickel-Based Alloys in The Paper IndustryDocument9 pagesSurface Characterisation and Crevice Corrosion Behaviour of Nickel-Based Alloys in The Paper IndustryANA LAURA BRAGA NASCIMENTONo ratings yet
- The Electrodeposition of Ternary Fe-Cr-Ni AlloysDocument28 pagesThe Electrodeposition of Ternary Fe-Cr-Ni AlloysMariianiita SalvatoreNo ratings yet
- 1-s2.0-S2238785422013734-mainDocument13 pages1-s2.0-S2238785422013734-mainGold SuganthNo ratings yet
- Materials Today: ProceedingsDocument6 pagesMaterials Today: ProceedingsSREEJITH S NAIRNo ratings yet
- ActaMat 51 ToroDocument12 pagesActaMat 51 Toroaltoro44No ratings yet
- Introduction to Stainless Steel OverviewDocument45 pagesIntroduction to Stainless Steel Overviewaladinmf1No ratings yet
- tmpEAB2 TMPDocument3 pagestmpEAB2 TMPFrontiersNo ratings yet
- Powder Metallurgy of Stainless Steels and Composites: A Review of Mechanical Alloying and Spark Plasma SinteringDocument20 pagesPowder Metallurgy of Stainless Steels and Composites: A Review of Mechanical Alloying and Spark Plasma Sinteringismail ismaNo ratings yet
- Zn-Mg-Al Corrosion analysis paperDocument12 pagesZn-Mg-Al Corrosion analysis paperrahul.meenaNo ratings yet
- Atmospheric corrosion of carbon steels and weathering steels in TaiwanDocument7 pagesAtmospheric corrosion of carbon steels and weathering steels in TaiwanSrikanth SrikantiNo ratings yet
- M. Bučko - 2014Document9 pagesM. Bučko - 2014terNo ratings yet
- Inhibitive Performance of A Rust Converter On Corrosion of Mild SteelDocument7 pagesInhibitive Performance of A Rust Converter On Corrosion of Mild SteelOmar MorteoNo ratings yet
- IJETR021817Document3 pagesIJETR021817erpublicationNo ratings yet
- Growth of Passive Films On Valve Metals and Their AlloysDocument9 pagesGrowth of Passive Films On Valve Metals and Their AlloysDjedili AmelNo ratings yet
- Journal of Materials Science & TechnologyDocument14 pagesJournal of Materials Science & TechnologyPATEL NIKUNJKUMAR JITENDRABHAINo ratings yet
- S Phase Surface Engineering of Fe CR Co CR and Ni CR Alloys - Opt PDFDocument35 pagesS Phase Surface Engineering of Fe CR Co CR and Ni CR Alloys - Opt PDFhernan nicolas tovar falonNo ratings yet
- 1 s2.0 S1526612517302542 MainDocument14 pages1 s2.0 S1526612517302542 Mainsudish mishraNo ratings yet
- Journal of Manufacturing Processes: Full Length ArticleDocument14 pagesJournal of Manufacturing Processes: Full Length ArticleSudish Jay MishraNo ratings yet
- Study of PlasmaDocument12 pagesStudy of PlasmaFerdinando Marco Rodrigues BorgesNo ratings yet
- A Study of Carbon Steels in Basic PittingDocument6 pagesA Study of Carbon Steels in Basic PittingSalem GarrabNo ratings yet
- Coatings 11 00517Document16 pagesCoatings 11 00517Domingo FernándezNo ratings yet
- Aging Behavior and Mechanical Properties of Maraging Steels in The Presence of Submicrocrystalline Laves Phase ParticlesDocument6 pagesAging Behavior and Mechanical Properties of Maraging Steels in The Presence of Submicrocrystalline Laves Phase ParticlesMarcelo de Freitas DantasNo ratings yet
- Nanomaterials 11 03298Document12 pagesNanomaterials 11 03298Liviu BadeaNo ratings yet
- Electro SynthesisDocument14 pagesElectro SynthesislapogadnanNo ratings yet
- Metals and Its AlloysDocument24 pagesMetals and Its AlloysrielcrizaldoNo ratings yet
- BF 02814824Document17 pagesBF 02814824Saiful ShokriNo ratings yet
- A Review of The Electrochemical Corrosion Behaviour of Iron AluminidesDocument10 pagesA Review of The Electrochemical Corrosion Behaviour of Iron AluminidesNgọc Minh LêNo ratings yet
- Cavitation Erosion of Aluminum Matrix Sintered Composite With AlN DispersoidsDocument5 pagesCavitation Erosion of Aluminum Matrix Sintered Composite With AlN DispersoidsRaluca FloreaNo ratings yet
- Krunal Vora ContentDocument14 pagesKrunal Vora ContentDevashish JoshiNo ratings yet
- Zhang 2012Document10 pagesZhang 2012Lan Đàm Thị PhươngNo ratings yet
- Microstructure and Mechanical Properties of A 5754 Aluminum Alloy Modified by SC and ZR AdditionsDocument9 pagesMicrostructure and Mechanical Properties of A 5754 Aluminum Alloy Modified by SC and ZR Additionshamdast64No ratings yet
- Engineering Failure AnalysisDocument19 pagesEngineering Failure AnalysisTimofey IgoninNo ratings yet
- Corrosion behaviour of Zn–Al–Mg coated steelDocument9 pagesCorrosion behaviour of Zn–Al–Mg coated steelAlex GarciaNo ratings yet
- Magnetic Properties of Stainless Steels at Room An PDFDocument9 pagesMagnetic Properties of Stainless Steels at Room An PDFJim SmithNo ratings yet
- Magnetic Properties of Stainless Steels at Room An PDFDocument9 pagesMagnetic Properties of Stainless Steels at Room An PDFJim SmithNo ratings yet
- The Use of Hydrogen in Shielding Gases Part I - Benefits and ConcernsDocument7 pagesThe Use of Hydrogen in Shielding Gases Part I - Benefits and ConcernsaauriscNo ratings yet
- Stainless Steel in The Food and Beverage IndustryDocument28 pagesStainless Steel in The Food and Beverage IndustryaauriscNo ratings yet
- Leaflet Corrosion of Arcelor MittalDocument6 pagesLeaflet Corrosion of Arcelor MittalKaushik PatelNo ratings yet
- Stainless Steels and Their PropertiesDocument45 pagesStainless Steels and Their PropertiesSH1961No ratings yet
- Leaflet Corrosion of Arcelor MittalDocument6 pagesLeaflet Corrosion of Arcelor MittalKaushik PatelNo ratings yet
- (Praxair) Argon and Hydrogen Gas BlendsDocument1 page(Praxair) Argon and Hydrogen Gas BlendsaauriscNo ratings yet
- The Use of Hydrogen in Shielding Gases Part II - Base Metal EffectsDocument6 pagesThe Use of Hydrogen in Shielding Gases Part II - Base Metal EffectsaauriscNo ratings yet
- The Use of Hydrogen in Shielding Gases Part II - Base Metal EffectsDocument6 pagesThe Use of Hydrogen in Shielding Gases Part II - Base Metal EffectsaauriscNo ratings yet
- (SOLGROUP) Enermix HydroDocument4 pages(SOLGROUP) Enermix HydroAlonso AurisNo ratings yet
- Stainless Steels and Their PropertiesDocument45 pagesStainless Steels and Their PropertiesSH1961No ratings yet
- (SOLGROUP) Enermix HydroDocument4 pages(SOLGROUP) Enermix HydroAlonso AurisNo ratings yet
- Linx® Shielding Gases: Lower Welding CostsDocument7 pagesLinx® Shielding Gases: Lower Welding CostsMahmoud shawkyNo ratings yet
- (AIR PRODUCTS) Inomaxx TIG - T1 ArH2 PDFDocument2 pages(AIR PRODUCTS) Inomaxx TIG - T1 ArH2 PDFaauriscNo ratings yet
- AusteniteDocument10 pagesAustenitekameshvvNo ratings yet
- Stainless SteelDocument8 pagesStainless SteelAlvaro Alexis Mendoza PradaNo ratings yet
- Atlas Technical Handbook of SS Rev Aug 2013Document49 pagesAtlas Technical Handbook of SS Rev Aug 2013timparker01No ratings yet
- Welding of Austenitic Stainless Steel - Job Knowledge 103Document3 pagesWelding of Austenitic Stainless Steel - Job Knowledge 103aauriscNo ratings yet
- Saddle Fusion.: ASTM F2620-11 The Heat-Fusion Procedures Covered in This Practice Are Socket Fusion, Butt Fusion, andDocument1 pageSaddle Fusion.: ASTM F2620-11 The Heat-Fusion Procedures Covered in This Practice Are Socket Fusion, Butt Fusion, andaauriscNo ratings yet
- Leaflet Corrosion of Arcelor MittalDocument6 pagesLeaflet Corrosion of Arcelor MittalKaushik PatelNo ratings yet
- Hellos PDFDocument1 pageHellos PDFaauriscNo ratings yet
- Stain Steel in Food IndustryDocument28 pagesStain Steel in Food IndustryJosé Luis Lustau Álvarez-CienfuegosNo ratings yet
- 1Document7 pages1aauriscNo ratings yet
- Astm D2657 - 07Document7 pagesAstm D2657 - 07dsalguero_2100% (2)
- 2Document9 pages2aauriscNo ratings yet
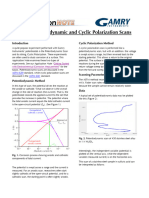
- Gamry - Poteniodynamic-and-Cyclic-PolarizationDocument3 pagesGamry - Poteniodynamic-and-Cyclic-PolarizationHenrique Ribeiro Piaggio CardosoNo ratings yet
- A Master's Guide To Ship's Piping 2nd EditionDocument42 pagesA Master's Guide To Ship's Piping 2nd Editiondassi99100% (1)
- Alloy 718Document16 pagesAlloy 718shyamNo ratings yet
- Case Study on Sulfide Stress Corrosion Cracking of an A216-WCC Wellhead Flow Control Valve BodyDocument12 pagesCase Study on Sulfide Stress Corrosion Cracking of an A216-WCC Wellhead Flow Control Valve Bodypinkan25No ratings yet
- The Metro Group Water Treatment HandbookDocument65 pagesThe Metro Group Water Treatment HandbookChoice OrganoNo ratings yet
- Everything You Need to Know About Corrosion ResistanceDocument12 pagesEverything You Need to Know About Corrosion ResistanceTal PeraltaNo ratings yet
- Fundamentals of Corrosion - Dr. Govind VagadiyaDocument14 pagesFundamentals of Corrosion - Dr. Govind VagadiyaDr. Govind VagadiyaNo ratings yet
- Failure - Mechanisms - of - C-Steels - API - 571 - .Xls - Filename UTF-8''Failure Mechanisms of C-Steels (API 571)Document100 pagesFailure - Mechanisms - of - C-Steels - API - 571 - .Xls - Filename UTF-8''Failure Mechanisms of C-Steels (API 571)أحمد صبحى100% (2)
- Duplex Stainless BrochureDocument13 pagesDuplex Stainless BrochurepramodtryNo ratings yet
- Corrosion of Structural SteelDocument6 pagesCorrosion of Structural SteelJaleel ClaasenNo ratings yet
- S 13 CRDocument6 pagesS 13 CRBardan RahmatanNo ratings yet
- Guide Line For SS Material in WATERDocument12 pagesGuide Line For SS Material in WATERAnonymous bHh1L1No ratings yet
- VT Inspect With BorescopeDocument54 pagesVT Inspect With BorescopeDodi SuhendraNo ratings yet
- CorrosionResistantAlloysintheOilandGasIndustrySelectionGuidelinesUpdate 10073Document12 pagesCorrosionResistantAlloysintheOilandGasIndustrySelectionGuidelinesUpdate 10073digecaNo ratings yet
- 5500 Centrismart ™ Operations Manual: Revised April 2004Document214 pages5500 Centrismart ™ Operations Manual: Revised April 2004Julio Cesar Romero HurtadoNo ratings yet
- Environmentally Assisted Cracking - Predictive Methods For Risk Assessment and Evaluation of Materials, Equipment, and Structures (ASTM Special Technical Publication, 1401) PDFDocument491 pagesEnvironmentally Assisted Cracking - Predictive Methods For Risk Assessment and Evaluation of Materials, Equipment, and Structures (ASTM Special Technical Publication, 1401) PDFNA Sleeper100% (1)
- Role of Nitrite Addition in Chloride Stress Corrosion Cracking of A Super Duplex Stainless SteelDocument5 pagesRole of Nitrite Addition in Chloride Stress Corrosion Cracking of A Super Duplex Stainless Steelherschel5No ratings yet
- Asset Preservation MethodsDocument10 pagesAsset Preservation MethodsLuck Luqe67% (3)
- NTNU TSA SprayingDocument89 pagesNTNU TSA SprayingAnnNo ratings yet
- CORROSION TYPES and PreventionDocument4 pagesCORROSION TYPES and PreventionHamid AlbashirNo ratings yet
- 9501JANDocument88 pages9501JANjroquel1084No ratings yet
- A-CableTray en PDFDocument294 pagesA-CableTray en PDFZainNo ratings yet
- Alloy 276 Spec SheetDocument3 pagesAlloy 276 Spec SheetermusatNo ratings yet
- Astm g34 - рск 2ххх и 7хххDocument8 pagesAstm g34 - рск 2ххх и 7хххIlyasNo ratings yet
- Pitting Corrosion Resistance of CA6NM and 410 Martensitic Stainless Steels in Various EnvironmentsDocument8 pagesPitting Corrosion Resistance of CA6NM and 410 Martensitic Stainless Steels in Various EnvironmentsAntonioNo ratings yet
- t38356 EngDocument7 pagest38356 Enghacene omarNo ratings yet
- Acom 3 2015Document13 pagesAcom 3 2015Joshua WalkerNo ratings yet
- DC Boiler Tube FailureDocument56 pagesDC Boiler Tube FailureDivyansh ChaturvediNo ratings yet
- Improve Glass-Lined ReactorDocument7 pagesImprove Glass-Lined ReactorRonaldo JanglinNo ratings yet