You might also like
- Assignment 1Document6 pagesAssignment 1Yi Hong LowNo ratings yet
- Investigating the Rate Constant for a Diffusion Controlled ReactionDocument5 pagesInvestigating the Rate Constant for a Diffusion Controlled ReactionJames HerdNo ratings yet
- Exercise 8 Kinetics of Hydrolysis of Ethyl AcetateDocument6 pagesExercise 8 Kinetics of Hydrolysis of Ethyl AcetatePalak BansalNo ratings yet
- MIL-PRF-7024E Calibrating Fluid SpecificationDocument10 pagesMIL-PRF-7024E Calibrating Fluid SpecificationkidseismicNo ratings yet
- CREII Module II Lecture 6 8Document79 pagesCREII Module II Lecture 6 8Aditya parasNo ratings yet
- CREII-Module-2 - Lecture 7 PDFDocument30 pagesCREII-Module-2 - Lecture 7 PDFshubhamNo ratings yet
- 3 Ley de Velocidad de ReacciónDocument44 pages3 Ley de Velocidad de ReacciónRonaldo Luis Guao BolañoNo ratings yet
- Tansition State Theory To Determine The Rate ConstantDocument6 pagesTansition State Theory To Determine The Rate Constant張智淵No ratings yet
- Fluid-Solid Catalytic Reactions: Rate-Limiting Step: Academic UseDocument11 pagesFluid-Solid Catalytic Reactions: Rate-Limiting Step: Academic UseshubhamNo ratings yet
- 3 Kinetika Reaksi 2018 (Part 1)Document13 pages3 Kinetika Reaksi 2018 (Part 1)Iqbal Al FuadyNo ratings yet
- Soran University Department of Chemical Engineering document on reaction types in Aspen HYSYSDocument24 pagesSoran University Department of Chemical Engineering document on reaction types in Aspen HYSYSAram Nasih MuhammadNo ratings yet
- CHPR4406 Reactions Lecture 1Document16 pagesCHPR4406 Reactions Lecture 1xx_aleksa_hrvatska_xxNo ratings yet
- Lecture 20: The Fixed Bed Catalytic Reactor: RXN RXNDocument10 pagesLecture 20: The Fixed Bed Catalytic Reactor: RXN RXNreddi ramuNo ratings yet
- Reactors1 16Document3 pagesReactors1 16Mourad kharbachNo ratings yet
- PDF DocumentDocument9 pagesPDF DocumentpalesaNo ratings yet
- TRK1 2013 Chapt 3 (Part 1)Document17 pagesTRK1 2013 Chapt 3 (Part 1)Yoel Dwi Putra GultomNo ratings yet
- Reactor EngineeringDocument19 pagesReactor EngineeringAlvine AyietaNo ratings yet
- Chemical Equilibrium ExplainedDocument11 pagesChemical Equilibrium ExplainedManu NathNo ratings yet
- Distillation Column in HYSYS: Kinetic Reaction ExampleDocument24 pagesDistillation Column in HYSYS: Kinetic Reaction ExampleAram Nasih MuhammadNo ratings yet
- 2019 Cre Ii L24-26Document32 pages2019 Cre Ii L24-26Aman PrasadNo ratings yet
- Multiphase Reactors: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocument49 pagesMultiphase Reactors: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiBikashGuptaNo ratings yet
- CREII-Module-I - Lecture 3Document27 pagesCREII-Module-I - Lecture 3Aditya parasNo ratings yet
- Lab 1.1 Theory - Reaction Kinetic Studies in A Batch ReactorDocument3 pagesLab 1.1 Theory - Reaction Kinetic Studies in A Batch ReactorCARL VINCENT SORIANONo ratings yet
- Rate Laws and StoichiometryDocument20 pagesRate Laws and StoichiometryAzreen AnisNo ratings yet
- Chemical Reactor Design: 7 Sem, B.Tech. Chemical EnggDocument20 pagesChemical Reactor Design: 7 Sem, B.Tech. Chemical EnggMohammad Ajaz DeshmukhNo ratings yet
- Lecture 11: Reactions in LiquidsDocument9 pagesLecture 11: Reactions in LiquidsGanesh Chandra BaroNo ratings yet
- Mass Transfer Coefficient ExplainedDocument37 pagesMass Transfer Coefficient ExplainednivedhithaNo ratings yet
- Topic 6 & 16: KineticsDocument63 pagesTopic 6 & 16: Kineticsapi-546066323No ratings yet
- Problem Set 6 SolutionDocument4 pagesProblem Set 6 SolutionRod De GuzmanNo ratings yet
- Chemistry Pre-U Chemistry Sem 1 Chap 5 PDFDocument85 pagesChemistry Pre-U Chemistry Sem 1 Chap 5 PDFJIANHUI0160% (1)
- Acre MSC Part 3 20 Feb 2014Document16 pagesAcre MSC Part 3 20 Feb 2014Salman HaroonNo ratings yet
- Lecture 18: Reactions at Solid SurfacesDocument12 pagesLecture 18: Reactions at Solid SurfacesGanesh Chandra BaroNo ratings yet
- Introduction & Overview To Chemical Reaction Engineering IIDocument12 pagesIntroduction & Overview To Chemical Reaction Engineering IIshubhamNo ratings yet
- Chapter 2b KineticsDocument11 pagesChapter 2b KineticsSankar SasmalNo ratings yet
- 4 Basics of ThermodynamicsDocument2 pages4 Basics of Thermodynamicsdeekshithasweety0No ratings yet
- Kinetics of Heterogeneous ReactionsDocument42 pagesKinetics of Heterogeneous ReactionsmikialeNo ratings yet
- Kinetics of Surface Catalysed RxnsDocument5 pagesKinetics of Surface Catalysed Rxnsnrj18No ratings yet
- CH2416 - BKC Revision NotesDocument2 pagesCH2416 - BKC Revision NotesJohnNo ratings yet
- Understanding Catalysis and Reactor DesignDocument43 pagesUnderstanding Catalysis and Reactor DesignLê MinhNo ratings yet
- CHM 111 Notes - 2021 - 2022Document19 pagesCHM 111 Notes - 2021 - 2022j9927091No ratings yet
- Boon PinDocument13 pagesBoon PinjayaprinaNo ratings yet
- CREII-Module-I - Lecture 4 PDFDocument34 pagesCREII-Module-I - Lecture 4 PDFshubhamNo ratings yet
- Eyring Equation ExplainedDocument5 pagesEyring Equation ExplainedBeaaaaNo ratings yet
- Kinetics, Rate Equations, RdsDocument10 pagesKinetics, Rate Equations, RdstomNo ratings yet
- Concentration Distributions in Different Phases at LaminarDocument23 pagesConcentration Distributions in Different Phases at LaminarHayder HusseinNo ratings yet
- Concentration Distributions in Different Phases at LaminarDocument23 pagesConcentration Distributions in Different Phases at LaminarHayder HusseinNo ratings yet
- Genchem Reviewer (2nd Grading)Document27 pagesGenchem Reviewer (2nd Grading)assassin1252005No ratings yet
- Chemical Dynamics Theoretical ToolsDocument54 pagesChemical Dynamics Theoretical ToolsRyan GoldenNo ratings yet
- Class XI Equilibrium NotesDocument8 pagesClass XI Equilibrium NoteseasaNo ratings yet
- Kinetics of Hydrolysis of Ethyl EsterDocument6 pagesKinetics of Hydrolysis of Ethyl EsterJawad AhmadNo ratings yet
- Assignment 1Document3 pagesAssignment 1NoranierahNohoNo ratings yet
- Pchem10e Solutions ch04Document10 pagesPchem10e Solutions ch04이호준No ratings yet
- Reaction Rates, GE 404Document6 pagesReaction Rates, GE 404mevadatinkalNo ratings yet
- Law of Mass ActionDocument13 pagesLaw of Mass ActionmadhujayarajNo ratings yet
- Week 2 Interphase Mass TransferDocument17 pagesWeek 2 Interphase Mass TransferKagendren AyanNo ratings yet
- Equilibrium: Q Explain General Expression?Document7 pagesEquilibrium: Q Explain General Expression?Aq RaufNo ratings yet
- Rates of ReactionDocument7 pagesRates of ReactionDoc_CrocNo ratings yet
- Measure Mass Transfer Coefficients Using an Electrochemical TechniqueDocument4 pagesMeasure Mass Transfer Coefficients Using an Electrochemical TechniqueusercmdmcNo ratings yet
- Working Guide to Vapor-Liquid Phase Equilibria CalculationsFrom EverandWorking Guide to Vapor-Liquid Phase Equilibria CalculationsRating: 5 out of 5 stars5/5 (1)
- Oxidation (Slide No 6-11) (22768)Document6 pagesOxidation (Slide No 6-11) (22768)shubhamNo ratings yet
- Emission FeeDocument5 pagesEmission FeeshubhamNo ratings yet
- CVMDocument20 pagesCVMkamitrNo ratings yet
- Pollution ControlDocument62 pagesPollution ControlshubhamNo ratings yet
- Tragedy of CommonsDocument2 pagesTragedy of CommonsshubhamNo ratings yet
- Diffusion example design and analysisDocument13 pagesDiffusion example design and analysisPaulinaNo ratings yet
- Emission FeeDocument5 pagesEmission FeeshubhamNo ratings yet
- 10 CL636Document19 pages10 CL636shubhamNo ratings yet
- Valuation MethodsDocument73 pagesValuation MethodsshubhamNo ratings yet
- 5.etch - Part 3 (2) (21613)Document31 pages5.etch - Part 3 (2) (21613)shubhamNo ratings yet
- CVMDocument20 pagesCVMkamitrNo ratings yet
- 8 CL636Document20 pages8 CL636shubhamNo ratings yet
- Environmental Economics Pollution Control: Mrinal Kanti DuttaDocument253 pagesEnvironmental Economics Pollution Control: Mrinal Kanti DuttashubhamNo ratings yet
- Class TCM CVMDocument34 pagesClass TCM CVMshubhamNo ratings yet
- CL 636 5 To 10Document211 pagesCL 636 5 To 10shubhamNo ratings yet
- NRR TransitionDocument6 pagesNRR TransitionshubhamNo ratings yet
- Imported CSV Data: Exercise 1Document17 pagesImported CSV Data: Exercise 1shubhamNo ratings yet
- Env DevDocument36 pagesEnv DevshubhamNo ratings yet
- Market Failure1Document37 pagesMarket Failure1shubhamNo ratings yet
- HS229 Course OutlineDocument10 pagesHS229 Course OutlineshubhamNo ratings yet
- NRR QuantifiedDocument8 pagesNRR QuantifiedshubhamNo ratings yet
- 3.lithography - Part 2 (3) (17508)Document33 pages3.lithography - Part 2 (3) (17508)shubhamNo ratings yet
- EnvEc OverviewDocument19 pagesEnvEc OverviewshubhamNo ratings yet
- 3.CL 636 - Photolithography - Part 1 (4) (14932)Document33 pages3.CL 636 - Photolithography - Part 1 (4) (14932)shubhamNo ratings yet
- Combined Hs229Document74 pagesCombined Hs229shubhamNo ratings yet
- MicroFabCH1 5Document168 pagesMicroFabCH1 5shubhamNo ratings yet
- Economics of Natural Resources: Resources, and (3) Resource EndowmentDocument31 pagesEconomics of Natural Resources: Resources, and (3) Resource EndowmentshubhamNo ratings yet
- Beol - CL 636 (2) (10759)Document37 pagesBeol - CL 636 (2) (10759)shubhamNo ratings yet
- CL 636 - Introduction - 1 (10500)Document11 pagesCL 636 - Introduction - 1 (10500)shubhamNo ratings yet
- SWC - Let's Get Into Research Intern!Document14 pagesSWC - Let's Get Into Research Intern!shubhamNo ratings yet
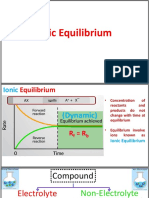
- Ionic Equilibrium ExplainedDocument14 pagesIonic Equilibrium Explained8842 AnuragNo ratings yet
- Free and Forced Convection Lab ReportDocument17 pagesFree and Forced Convection Lab ReportKamal GamalNo ratings yet
- Science Form 1-Chapter 1Document53 pagesScience Form 1-Chapter 1Mohamad TarmiziNo ratings yet
- Is - 14845Document17 pagesIs - 14845mechftpNo ratings yet
- Shielded Metal Arc Welding GuideDocument4 pagesShielded Metal Arc Welding GuideLloyd AlmonteNo ratings yet
- Xylem FLYGT Model Mixed FlowDocument12 pagesXylem FLYGT Model Mixed FlowEdilberto AlegatoNo ratings yet
- Varicel Ii MH With Intersept: Iaq Engineered High Efficiency Mini-Pleat Filter With Metal Header and Cell SidesDocument2 pagesVaricel Ii MH With Intersept: Iaq Engineered High Efficiency Mini-Pleat Filter With Metal Header and Cell SidesdbricchiNo ratings yet
- Crystal Defect - Planar Defects WNPDocument33 pagesCrystal Defect - Planar Defects WNPNaufal HNo ratings yet
- Painting Standard For DCVS Ombilin 20190926Document5 pagesPainting Standard For DCVS Ombilin 20190926fendiNo ratings yet
- Single Bond Universal - TPP - CEE-LA-APAC - PDFDocument40 pagesSingle Bond Universal - TPP - CEE-LA-APAC - PDFNabel ramiNo ratings yet
- Alloying Elements of Steels and PropertiesDocument3 pagesAlloying Elements of Steels and PropertiesdaimaheshNo ratings yet
- Curzard's Worm CYOADocument9 pagesCurzard's Worm CYOAMortuus EstNo ratings yet
- Salt and SugarDocument9 pagesSalt and Sugarapi-384186386No ratings yet
- Perlus H 32 Hydraulic Oil SpecsDocument2 pagesPerlus H 32 Hydraulic Oil SpecsNermine NasrNo ratings yet
- Arash Golchin - Komplett IIDocument118 pagesArash Golchin - Komplett IIAmber WilliamsNo ratings yet
- Jurnal Hama Kumbang Logong Pada BerasDocument12 pagesJurnal Hama Kumbang Logong Pada BerasRenaldo MoontriNo ratings yet
- Comsol y Ondas UltrasonicasDocument7 pagesComsol y Ondas UltrasonicasCelesteCebedioNo ratings yet
- Femtosecond Laser PDFDocument2 pagesFemtosecond Laser PDFVirginiaNo ratings yet
- Ch. 17Document11 pagesCh. 17amedawg3No ratings yet
- Urea 25 22 2 20030603Document4 pagesUrea 25 22 2 20030603Ghulam AhmadNo ratings yet
- Catalogue PageDocument3 pagesCatalogue PageDhairyaNo ratings yet
- Silo 9 Boulders Investigation ReportDocument20 pagesSilo 9 Boulders Investigation ReportWaka OngetiNo ratings yet
- SS207 Oil Pressure Sensor Product SpecsDocument12 pagesSS207 Oil Pressure Sensor Product Specsmarvin17No ratings yet
- AHU Drain CalculationDocument1 pageAHU Drain Calculationeldosrajan100% (1)
- Ruegg Catalogo Info Etechn eDocument36 pagesRuegg Catalogo Info Etechn eTiago MatosNo ratings yet
- C2 Representing Reactions HigherDocument11 pagesC2 Representing Reactions HigherdownendscienceNo ratings yet
- Medical Students Guide to Drug DispensingDocument40 pagesMedical Students Guide to Drug DispensingJIBRIL AHMEDNo ratings yet
- ObjectivesDocument3 pagesObjectivesSayeRuzanaNo ratings yet
- German ATV DVWK A 168E Corrosion of Wastewater Systems Wastewater 1998 PDFDocument51 pagesGerman ATV DVWK A 168E Corrosion of Wastewater Systems Wastewater 1998 PDFJosip Medved100% (1)