You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Metal Gear Solid - Characters Profile and TimelineDocument65 pagesMetal Gear Solid - Characters Profile and Timelinekrevorkian1653100% (1)
- Question BankDocument9 pagesQuestion Bankpragash100% (1)
- Ancient Indian ArchitectureDocument86 pagesAncient Indian ArchitectureRishika100% (2)
- V Upashama PrakaranamDocument488 pagesV Upashama PrakaranamantiX LinuxNo ratings yet
- Sir Syed Ahmed Khan and Two Nation TheoryDocument11 pagesSir Syed Ahmed Khan and Two Nation TheoryZeeshan Saeed100% (5)
- The Life of DadajiDocument87 pagesThe Life of DadajiMike Magee100% (1)
- Atypical (Dysplastic) Nevus: Rainer Hofmann-Wellenhof and H. Peter SoyerDocument10 pagesAtypical (Dysplastic) Nevus: Rainer Hofmann-Wellenhof and H. Peter SoyerSuzana SoozyNo ratings yet
- A Comparative Study of Proliferative Nodules and Lethal Melanomas in Congenital Nevi From ChildrenDocument11 pagesA Comparative Study of Proliferative Nodules and Lethal Melanomas in Congenital Nevi From ChildrenSuzana SoozyNo ratings yet
- Congenital Melanocytic Nevi: ReviewDocument7 pagesCongenital Melanocytic Nevi: ReviewSuzana SoozyNo ratings yet
- Treatment Options For Giant Congenital Naevi: C. M. LawrenceDocument5 pagesTreatment Options For Giant Congenital Naevi: C. M. LawrenceSuzana SoozyNo ratings yet
- Dysplastic Nevi in Children: Special SymposiumDocument18 pagesDysplastic Nevi in Children: Special SymposiumSuzana SoozyNo ratings yet
- JA A D: M CAD ErmatolDocument2 pagesJA A D: M CAD ErmatolSuzana SoozyNo ratings yet
- Nast 2017Document13 pagesNast 2017Suzana SoozyNo ratings yet
- Dermoscopic Analysis of Nevus Sebaceus of Jadassohn: A Study of 13 CasesDocument9 pagesDermoscopic Analysis of Nevus Sebaceus of Jadassohn: A Study of 13 CasesSuzana SoozyNo ratings yet
- Abboud 2017Document5 pagesAbboud 2017Suzana SoozyNo ratings yet
- Advice After Minor Skin Surgery or Cautery Using Local AnaestheticDocument7 pagesAdvice After Minor Skin Surgery or Cautery Using Local AnaestheticSuzana SoozyNo ratings yet
- RETICULOIDUL ACTINIC Ro 320Document7 pagesRETICULOIDUL ACTINIC Ro 320Suzana SoozyNo ratings yet
- Sindromul Metabolic La Pacienþii Cu Lichen Plan Oral Metabolic Syndrome in Patients With Oral Lichen PlanusDocument7 pagesSindromul Metabolic La Pacienþii Cu Lichen Plan Oral Metabolic Syndrome in Patients With Oral Lichen PlanusSuzana SoozyNo ratings yet
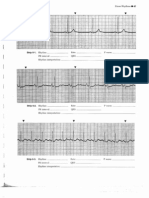
- Modele de EKGDocument43 pagesModele de EKGSuzana Soozy100% (1)
- Analytic Solutions of A Two Dimensional Rectangular Heat Equation PDFDocument5 pagesAnalytic Solutions of A Two Dimensional Rectangular Heat Equation PDFreff1694No ratings yet
- Councillor Danny Thorpe: Leader, Royal Borough of GreenwichDocument2 pagesCouncillor Danny Thorpe: Leader, Royal Borough of GreenwichDr-Syed Ali TarekNo ratings yet
- Global Business CH 2 2-MergedDocument58 pagesGlobal Business CH 2 2-MergedJace gunnerNo ratings yet
- (267.) SWOT - Cruise Industry & CarnivalDocument2 pages(267.) SWOT - Cruise Industry & CarnivalBilly Julius Gestiada100% (1)
- Classroom EtiquetteDocument1 pageClassroom EtiquetteJerrold MangaoNo ratings yet
- Hostel Standard and Safety MeasuresDocument16 pagesHostel Standard and Safety MeasuresSadiya IqbalNo ratings yet
- 1620 Ultra High Speed: Continuous Ink Jet PrinterDocument8 pages1620 Ultra High Speed: Continuous Ink Jet PrinterRudiNo ratings yet
- Particulate Ow at The Mouth of The Soummam Watershed (Algeria)Document9 pagesParticulate Ow at The Mouth of The Soummam Watershed (Algeria)bougheraraNo ratings yet
- Conscious Sedation PaediatricsDocument44 pagesConscious Sedation PaediatricsReeta TaxakNo ratings yet
- Artificial RainfallDocument20 pagesArtificial Rainfallsurajsingh100% (3)
- CAT LOGO ALPINESTARS OFF-ROAD 2021Document73 pagesCAT LOGO ALPINESTARS OFF-ROAD 2021Carla ArcosNo ratings yet
- Dr. Shikha Baskar: CHE154: Physical Chemistry For HonorsDocument22 pagesDr. Shikha Baskar: CHE154: Physical Chemistry For HonorsnishitsushantNo ratings yet
- Ol7 SecurityDocument84 pagesOl7 Securityneaman_ahmedNo ratings yet
- English Slide Un SMK 1819Document93 pagesEnglish Slide Un SMK 1819EkhaSoemarnoNo ratings yet
- The Awakened Goat: Playable RaceDocument2 pagesThe Awakened Goat: Playable RacePJ FlandersNo ratings yet
- Antenna Mini ProjectDocument65 pagesAntenna Mini ProjectHassan MehsenNo ratings yet
- PHP - Google Maps - How To Get GPS Coordinates For AddressDocument4 pagesPHP - Google Maps - How To Get GPS Coordinates For AddressAureliano DuarteNo ratings yet
- 01 IKSP and Environmental MovementsDocument12 pages01 IKSP and Environmental MovementsGlister Diadem DolleraNo ratings yet
- The Human Body: Winston S. Abena, RMT, MD, DPASMAPDocument51 pagesThe Human Body: Winston S. Abena, RMT, MD, DPASMAPMinelli ArgonzaNo ratings yet
- Cosep v. People PDFDocument8 pagesCosep v. People PDFAlyssa Fatima Guinto SaliNo ratings yet
- RNR150 Syllabus1Document14 pagesRNR150 Syllabus1Shaun SarvisNo ratings yet
- Timely Hints OctoberDocument5 pagesTimely Hints OctoberDane McDonaldNo ratings yet
- ExcaDrill 45A DF560L DatasheetDocument2 pagesExcaDrill 45A DF560L DatasheetИгорь ИвановNo ratings yet