You might also like
- Research in Physiopathology As Basis of Guided Chemotherapy With Special Application To Cancer Emanuel Revici, 1961Document806 pagesResearch in Physiopathology As Basis of Guided Chemotherapy With Special Application To Cancer Emanuel Revici, 1961wxcvbnnbvcxwNo ratings yet
- Cancer AwarenessDocument20 pagesCancer AwarenessArul Nambi Ramanujam100% (3)
- Clinical Microbiology and Infection: Original ArticleDocument6 pagesClinical Microbiology and Infection: Original ArticleAkira Masumi100% (1)
- 08 Pathology Orthobullets2017 PDFDocument189 pages08 Pathology Orthobullets2017 PDFdr_shafiq100% (1)
- Dr. Yoseph - The Role of Radiotherapy in The Management of Ameloblastoma and Ameloblastic CarcinomaDocument10 pagesDr. Yoseph - The Role of Radiotherapy in The Management of Ameloblastoma and Ameloblastic CarcinomaOnkologi Radiasi Angkatan 23No ratings yet
- Supplement: Workshop On Radiation Dosimetry: Basic Technologies, Medical Applications, Environmental ApplicationsDocument86 pagesSupplement: Workshop On Radiation Dosimetry: Basic Technologies, Medical Applications, Environmental ApplicationsLiviu CraciunNo ratings yet
- Thesis Presentation - For UniDocument36 pagesThesis Presentation - For UniDani Simón ColomarNo ratings yet
- Construction, Design and Comissioning of MLC ForDocument74 pagesConstruction, Design and Comissioning of MLC ForSAlonii ChawlaNo ratings yet
- ILRT Dr. Sarbani-1 PDFDocument48 pagesILRT Dr. Sarbani-1 PDFdurgesh kumar100% (1)
- Tumor OITE - 2012 2013 2014Document254 pagesTumor OITE - 2012 2013 2014ICH Khuy100% (1)
- Brain and Spinal Tumors of ChildhoodDocument560 pagesBrain and Spinal Tumors of ChildhoodmabidsaleemNo ratings yet
- Radiation Safety Test Aramco Sample Q A 2 PDFDocument22 pagesRadiation Safety Test Aramco Sample Q A 2 PDFnaoufel1706100% (1)
- Oncology Nursing Part 3Document23 pagesOncology Nursing Part 3fleur harrisonNo ratings yet
- Monaco 5.10 Training GuideDocument1,194 pagesMonaco 5.10 Training GuideMircea Ciocaltei100% (1)
- Diagnostic Approach to Musculoskeletal TumorsDocument25 pagesDiagnostic Approach to Musculoskeletal TumorsFatini ChokNo ratings yet
- Common Childhood Bone Cancer GuideDocument18 pagesCommon Childhood Bone Cancer GuideMohammed SalimNo ratings yet
- Pediatric Osteosarcoma: Pearls and PitfallsDocument18 pagesPediatric Osteosarcoma: Pearls and PitfallsNicolas Cuellar FernandezNo ratings yet
- Ew WikiDocument8 pagesEw WikidesyonkNo ratings yet
- Bone TumorsDocument43 pagesBone TumorsIsaac MwangiNo ratings yet
- Bone TumorsDocument8 pagesBone TumorsHikari AoiNo ratings yet
- Osteosarcoma Diagnosis and TreatmentDocument6 pagesOsteosarcoma Diagnosis and TreatmentBeladiena Citra SiregarNo ratings yet
- Beird Hannah C Osteosarcoma 2022 12Document19 pagesBeird Hannah C Osteosarcoma 2022 12najibNo ratings yet
- Ewing SarkomaDocument30 pagesEwing SarkomaullyNo ratings yet
- Primary Osseous Tumors of The Pediatric Spinal ColumnDocument14 pagesPrimary Osseous Tumors of The Pediatric Spinal ColumnthanhdhyscribdNo ratings yet
- Asuhan Keperawatan Medikal Bedah Dengan Tumor TulangDocument30 pagesAsuhan Keperawatan Medikal Bedah Dengan Tumor TulangYeni SeptianiNo ratings yet
- Sarcoma de EdwingDocument14 pagesSarcoma de EdwingricardoNo ratings yet
- Osteosarcoma Mini-SymposiumDocument11 pagesOsteosarcoma Mini-SymposiumJawad KhanNo ratings yet
- Common Musculoskeletal Tumors of ChildhoodDocument13 pagesCommon Musculoskeletal Tumors of ChildhoodMari CherNo ratings yet
- Jurnal Pendahuluan MutaDocument23 pagesJurnal Pendahuluan MutaMuta MimahNo ratings yet
- Small Round Cell Tumors of Soft Tissue and BoneDocument13 pagesSmall Round Cell Tumors of Soft Tissue and BoneAdriana Gabriela Ugarte MacíasNo ratings yet
- OsteosarcomaDocument12 pagesOsteosarcomaBrigdite Brii Zuleta CortezNo ratings yet
- Lec. 8 Malignant Bone TumorsDocument17 pagesLec. 8 Malignant Bone Tumorsنور كاضمNo ratings yet
- Jurnal 0steosarcoma 2Document27 pagesJurnal 0steosarcoma 2Rika Irena DwiputriNo ratings yet
- Hyl 142Document11 pagesHyl 142Stefania CristinaNo ratings yet
- Osteo Sarko Ma 1Document10 pagesOsteo Sarko Ma 1merizNo ratings yet
- Low-Grade Central Osteosarcoma Versus Fibrous Dysplasia: Carrie Y. Inwards, MDDocument6 pagesLow-Grade Central Osteosarcoma Versus Fibrous Dysplasia: Carrie Y. Inwards, MDManuelGonzalezGaitanoNo ratings yet
- Chapter 455 Retinoblastoma Retinoblastoma Charles B. Pratt: PathologyDocument4 pagesChapter 455 Retinoblastoma Retinoblastoma Charles B. Pratt: PathologyEbook Kedokteran Bahan KuliahNo ratings yet
- New Horizons in The Treatment of Osteosarcoma: Review ArticleDocument11 pagesNew Horizons in The Treatment of Osteosarcoma: Review ArticleRucelia Michiko PiriNo ratings yet
- Osteosarcoma: Pathology, Staging and Management: Eview RticleDocument10 pagesOsteosarcoma: Pathology, Staging and Management: Eview RticleAditya Rahman RYNo ratings yet
- Oral Pathology Lec - 1Document11 pagesOral Pathology Lec - 1مصطفى محمدNo ratings yet
- Cureus 0012 00000008418Document10 pagesCureus 0012 00000008418فرجني موغNo ratings yet
- Piis1319453413000660 PDFDocument10 pagesPiis1319453413000660 PDFZhakiAlAsrorNo ratings yet
- Non-Odontogenic Tumor (Lecture)Document64 pagesNon-Odontogenic Tumor (Lecture)shabeelpn86% (7)
- Ewing's SarcomaDocument10 pagesEwing's SarcomaVerli Fajriati NofliNo ratings yet
- Session 9 MalignanciesDocument92 pagesSession 9 MalignanciesZNo ratings yet
- Imaging Features of Bone Tumors on X-rays and MRIDocument15 pagesImaging Features of Bone Tumors on X-rays and MRIPedro Gabriel Quispe LópezNo ratings yet
- Articulo 1Document17 pagesArticulo 1Andrea NeriaNo ratings yet
- Spindle Cell LesionsDocument8 pagesSpindle Cell LesionsdrmanishsharmaNo ratings yet
- Bone tm3Document57 pagesBone tm3ZakiyahulfahdwNo ratings yet
- Sarcoma: StatisticsDocument1 pageSarcoma: StatisticsAngelica LupuNo ratings yet
- 12 Bone and Soft Tissue Sarcomas 200609 v2Document58 pages12 Bone and Soft Tissue Sarcomas 200609 v2dr. Ahmad MuhsininNo ratings yet
- 2017 Diagnostic Methods For Detection of Bone MetsDocument6 pages2017 Diagnostic Methods For Detection of Bone MetsgammasharkNo ratings yet
- Surgical Oncology: SciencedirectDocument8 pagesSurgical Oncology: SciencedirectMIGUEL MORENONo ratings yet
- Multiple Myeloma: EtiologyDocument3 pagesMultiple Myeloma: EtiologyJacob ChristianNo ratings yet
- ID NoneDocument9 pagesID NoneNur AnisaNo ratings yet
- Reviews: Advancing Therapy For OsteosarcomaDocument16 pagesReviews: Advancing Therapy For OsteosarcomaElleNo ratings yet
- Clinical Features and Diagnosis of Neoplastic Epidural Spinal Cord Compression - UpToDateDocument19 pagesClinical Features and Diagnosis of Neoplastic Epidural Spinal Cord Compression - UpToDatePrincess Jeanne Roque GairanodNo ratings yet
- 10.1038@s41572 020 00216 3Document28 pages10.1038@s41572 020 00216 3Jessica CampoNo ratings yet
- Ewings SarcomaDocument9 pagesEwings SarcomaElvisNo ratings yet
- Cancers 14 03503Document28 pagesCancers 14 03503ZhouChuQiaoNo ratings yet
- Primary Chest Wall TumorsDocument13 pagesPrimary Chest Wall Tumorsmhany12345No ratings yet
- 6 Pediatric Odontogenic Tumors. Oral and Maxillofacial Surgery Clinics of North AmericaDocument14 pages6 Pediatric Odontogenic Tumors. Oral and Maxillofacial Surgery Clinics of North AmericaalexandreNo ratings yet
- Inherited Genetic Syndromes. Certain Rare Genetic Syndromes Passed ThroughDocument3 pagesInherited Genetic Syndromes. Certain Rare Genetic Syndromes Passed ThroughOlivia AngNo ratings yet
- Small Round Cell TumorsDocument131 pagesSmall Round Cell TumorschinnnababuNo ratings yet
- Lytic Bone Lesions - StatPearls - NCBI BookshelfDocument9 pagesLytic Bone Lesions - StatPearls - NCBI Bookshelfjuan ricardo carvajal alvaradoNo ratings yet
- SC Compression, Neurol Clinics, 2-03Document20 pagesSC Compression, Neurol Clinics, 2-03Jaime H. Soto LugoNo ratings yet
- Session 9 Bone DiseasesDocument70 pagesSession 9 Bone DiseasesZNo ratings yet
- Neuroendocrine Tumors: Surgical Evaluation and ManagementFrom EverandNeuroendocrine Tumors: Surgical Evaluation and ManagementJordan M. CloydNo ratings yet
- Basal Cell Carcinoma: Advances in Treatment and ResearchFrom EverandBasal Cell Carcinoma: Advances in Treatment and ResearchMichael R. MigdenNo ratings yet
- Target Volume Delineation for Pediatric CancersFrom EverandTarget Volume Delineation for Pediatric CancersStephanie A. TerezakisNo ratings yet
- Simple Professional Virtual Meeting by SlidesgoDocument45 pagesSimple Professional Virtual Meeting by SlidesgoAkira MasumiNo ratings yet
- Art Subject For Elementary - 3rd Grade - Performing Arts by SlidesgoDocument56 pagesArt Subject For Elementary - 3rd Grade - Performing Arts by SlidesgoAkira MasumiNo ratings yet
- Sports and Physical Activity During COVID-19 by SlidesgoDocument56 pagesSports and Physical Activity During COVID-19 by SlidesgoBeldia Cáceres CataldoNo ratings yet
- Simple Professional Virtual Meeting by SlidesgoDocument45 pagesSimple Professional Virtual Meeting by SlidesgoAkira MasumiNo ratings yet
- Lessons Learned About COVID-19 by SlidesgoDocument34 pagesLessons Learned About COVID-19 by SlidesgoAkira MasumiNo ratings yet
- Sports and Physical Activity During COVID-19 by SlidesgoDocument56 pagesSports and Physical Activity During COVID-19 by SlidesgoBeldia Cáceres CataldoNo ratings yet
- Febrile Seizures v19 WebDocument1 pageFebrile Seizures v19 WebAkira MasumiNo ratings yet
- Lessons Learned About COVID-19 by SlidesgoDocument34 pagesLessons Learned About COVID-19 by SlidesgoAkira MasumiNo ratings yet
- COVID-19 Vaccine Breakthrough - Case Investigation and Reporting by SlidesgoDocument53 pagesCOVID-19 Vaccine Breakthrough - Case Investigation and Reporting by SlidesgoAkira MasumiNo ratings yet
- COVID-19 Vaccine Breakthrough - Case Investigation and Reporting by SlidesgoDocument53 pagesCOVID-19 Vaccine Breakthrough - Case Investigation and Reporting by SlidesgoAkira MasumiNo ratings yet
- Azithromycin and Cipro Oxacin Inhibit Interleukin-Secretion Without Disrupting Human Sinonasal Epithelial Integrity in VitroDocument8 pagesAzithromycin and Cipro Oxacin Inhibit Interleukin-Secretion Without Disrupting Human Sinonasal Epithelial Integrity in VitroAkira MasumiNo ratings yet
- Art Subject For Elementary - 3rd Grade - Performing Arts by SlidesgoDocument56 pagesArt Subject For Elementary - 3rd Grade - Performing Arts by SlidesgoAkira MasumiNo ratings yet
- Medicina: Inflammation and Endotyping in Chronic Rhinosinusitis-A Paradigm ShiftDocument13 pagesMedicina: Inflammation and Endotyping in Chronic Rhinosinusitis-A Paradigm ShiftAkira MasumiNo ratings yet
- Imaging in The Jaundiced Child PDFDocument14 pagesImaging in The Jaundiced Child PDFRahma Puji LestariNo ratings yet
- HHS Public Access: Chronic Rhinosinusitis Without Nasal PolypsDocument16 pagesHHS Public Access: Chronic Rhinosinusitis Without Nasal PolypsAnnisa RahmadhaniaNo ratings yet
- Traumatologi 2015Document5 pagesTraumatologi 2015Jhun EjuNo ratings yet
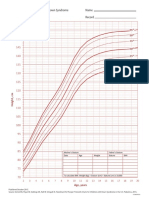
- DS Girls Height 2to20yearsDocument1 pageDS Girls Height 2to20yearsAkira MasumiNo ratings yet
- Inflammatory Biomarkers During Bacterial Acute RhinosinusitisDocument6 pagesInflammatory Biomarkers During Bacterial Acute RhinosinusitisAkira MasumiNo ratings yet
- DS Boys HeadCirc Birthto36moDocument1 pageDS Boys HeadCirc Birthto36moPeregrine Albertus Ricco AzaliNo ratings yet
- DS Boys HeadCirc 2-20yearsDocument1 pageDS Boys HeadCirc 2-20yearsInayahMaulaNo ratings yet
- Association Between Hypertensive Disorders of Pregnancy and Later Risk of CardiomyopathyDocument8 pagesAssociation Between Hypertensive Disorders of Pregnancy and Later Risk of CardiomyopathyPujiyono SuwadiNo ratings yet
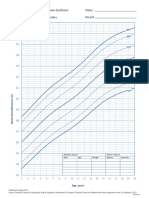
- Ds Girls Length Birthto36moDocument1 pageDs Girls Length Birthto36moWahyu DinataNo ratings yet
- The Association of Leptospermum Honey With Cytokine Expression in The Sinonasal Epithelium of Chronic Rhinosinusitis PatientsDocument7 pagesThe Association of Leptospermum Honey With Cytokine Expression in The Sinonasal Epithelium of Chronic Rhinosinusitis PatientsAkira MasumiNo ratings yet
- Effect of High Add Power, Medium Add Power, or Single-VisionDocument10 pagesEffect of High Add Power, Medium Add Power, or Single-VisionAkira MasumiNo ratings yet
- Departemen Biostatistika Dan Kependudukan, Fakultas Kesehatan Masyarakat Kampus C UNAIR Mulyorejo Alamat Korespondensi: Alvita Brilliana R.ADocument11 pagesDepartemen Biostatistika Dan Kependudukan, Fakultas Kesehatan Masyarakat Kampus C UNAIR Mulyorejo Alamat Korespondensi: Alvita Brilliana R.AHeiddy Ch SumampouwNo ratings yet
- Febrile Seizures v19 WebDocument1 pageFebrile Seizures v19 WebAkira MasumiNo ratings yet
- Orthopaedic Tumors and Masses - Clinical GateDocument9 pagesOrthopaedic Tumors and Masses - Clinical GateAkira MasumiNo ratings yet
- The Association of Leptospermum Honey With Cytokine Expression in The Sinonasal Epithelium of Chronic Rhinosinusitis PatientsDocument7 pagesThe Association of Leptospermum Honey With Cytokine Expression in The Sinonasal Epithelium of Chronic Rhinosinusitis PatientsAkira MasumiNo ratings yet
- Pelvis Lab 1 1Document13 pagesPelvis Lab 1 1api-632146706No ratings yet
- High Grade Gliomas:: Pathogenesis, Management and PrognosisDocument6 pagesHigh Grade Gliomas:: Pathogenesis, Management and Prognosisimanuela sinkyNo ratings yet
- Juvenile Nasopharyngeal AngiofibromaDocument6 pagesJuvenile Nasopharyngeal AngiofibromaSivaneasan KandiahNo ratings yet
- Liver CancerDocument28 pagesLiver CancerHealth Education Library for PeopleNo ratings yet
- The Radiological Accident at The Irradiation Facility in NesvizhDocument88 pagesThe Radiological Accident at The Irradiation Facility in NesvizhcavrisNo ratings yet
- Soft Tissue TumoursDocument27 pagesSoft Tissue TumoursNick ManikNo ratings yet
- Monaco Treatment Planning Enhances Departmental EfficienciesDocument9 pagesMonaco Treatment Planning Enhances Departmental Efficienciesricky rdnNo ratings yet
- Nasopharyngeal AngiofibromaDocument51 pagesNasopharyngeal Angiofibromakamal saudNo ratings yet
- Fefg EBM 2017 18 23 MarchDocument26 pagesFefg EBM 2017 18 23 MarchShivam AgarwalNo ratings yet
- 19181111Document104 pages19181111Jennifer HaasNo ratings yet
- Society of Radiographers - PDFDocument9 pagesSociety of Radiographers - PDFOsama AhmedNo ratings yet
- The Lost Tomb - Baritone TCDocument2 pagesThe Lost Tomb - Baritone TCDavid Apraez LeytonNo ratings yet
- Radiation Effects On The SkinDocument30 pagesRadiation Effects On The Skinsuryo111No ratings yet
- Radiation Damage in Biomolecules and CellsDocument400 pagesRadiation Damage in Biomolecules and CellsStephanie ChristineNo ratings yet
- Olfactory Neuroblastoma UP TO DATEDocument22 pagesOlfactory Neuroblastoma UP TO DATEZuleibis Melean LopezNo ratings yet
- Catalogo CIVCODocument94 pagesCatalogo CIVCORicardo Aguilar100% (1)
- Jeong and Jeong, 2018Document13 pagesJeong and Jeong, 2018yanuarNo ratings yet
- Budwig Guide 2019 01Document32 pagesBudwig Guide 2019 01Sasha TucakovicNo ratings yet
- Physica Medica: M.S. Herrera, S.J. González, D.M. Minsky, A.J. KreinerDocument11 pagesPhysica Medica: M.S. Herrera, S.J. González, D.M. Minsky, A.J. KreinerTaufiqur Rohman ChimeraNo ratings yet