You might also like
- Recent Advances in Ketene ChemistryDocument76 pagesRecent Advances in Ketene ChemistrylazersteveNo ratings yet
- Shuai 2020Document5 pagesShuai 2020Virat KohNo ratings yet
- Strategy in Synthesis-08Document34 pagesStrategy in Synthesis-08fjewafhjeashfeshfNo ratings yet
- A Study On Organic Synthesis Via KeteneDocument13 pagesA Study On Organic Synthesis Via Ketenesiyengar1447No ratings yet
- CHM102 20202021 Soln @anbc Academics Unit @blacts TutorialsDocument8 pagesCHM102 20202021 Soln @anbc Academics Unit @blacts Tutorialsbalikisolayemi2005No ratings yet
- 2 - H-NMRDocument38 pages2 - H-NMRahmed mohamedNo ratings yet
- Chemistry Insight Review TestDocument7 pagesChemistry Insight Review TestdivikjainvmcNo ratings yet
- Practice Exam 1Document5 pagesPractice Exam 1ManuelPauloAcogidoNo ratings yet
- Chen 2012Document3 pagesChen 2012Jaydeep MokariyaNo ratings yet
- Jumaah CarragenanoDocument8 pagesJumaah CarragenanoOrlandoCialliNo ratings yet
- JuvabioneDocument13 pagesJuvabionePreeti Yadav100% (1)
- 2016 Chemistry H1 JC2 Anglo Chinese Junior CollegeDocument48 pages2016 Chemistry H1 JC2 Anglo Chinese Junior CollegeLinn TanNo ratings yet
- Carbon Dioxide 1687608695Document3 pagesCarbon Dioxide 1687608695bayoalpha23No ratings yet
- Photoelectrochemical Water Splitting in Separate Oxygen and Hydrogen CellsDocument7 pagesPhotoelectrochemical Water Splitting in Separate Oxygen and Hydrogen Cellskhan47pkNo ratings yet
- AntibioticsDocument102 pagesAntibioticsKasim UmarNo ratings yet
- Organic Chemistry - HybridizationDocument24 pagesOrganic Chemistry - Hybridizationyamkela060.comNo ratings yet
- Question Bank On Unit IVDocument2 pagesQuestion Bank On Unit IVMowleen ArmstrongNo ratings yet
- 2010 November Understanding The VRFBDocument27 pages2010 November Understanding The VRFBljubisa milosevicNo ratings yet
- Of With Neutral N,: Host-Guest Structure XH2 C)Document10 pagesOf With Neutral N,: Host-Guest Structure XH2 C)Ahmad UsmanNo ratings yet
- Enantioselective (3+3) Atroposelective Annulation Catalyzed by N-Heterocyclic CarbenesDocument10 pagesEnantioselective (3+3) Atroposelective Annulation Catalyzed by N-Heterocyclic CarbenesKatrin MarchenkoNo ratings yet
- Free-Radical-Initiated Coupling Reaction of Alcohols and Alkynes: Not C-O But C-C Bond FormationDocument3 pagesFree-Radical-Initiated Coupling Reaction of Alcohols and Alkynes: Not C-O But C-C Bond FormationMohammad WahyuNo ratings yet
- Alkoxy Radicals: Scheme 1Document17 pagesAlkoxy Radicals: Scheme 1Baban BaidyaNo ratings yet
- Chemistry Assignment 1 - Physical StudentsDocument3 pagesChemistry Assignment 1 - Physical StudentsWebby ZimbaNo ratings yet
- Problems 3 BDocument2 pagesProblems 3 BLngNo ratings yet
- PJ 201431Document8 pagesPJ 201431Abdulrahman SaadNo ratings yet
- Structural Organisation of Proteins: Primary StructureDocument41 pagesStructural Organisation of Proteins: Primary StructureanaNo ratings yet
- Accepted Manuscript: Catalysis TodayDocument25 pagesAccepted Manuscript: Catalysis TodayGonzalo BenavidesNo ratings yet
- Bioorganic & Medicinal Chemistry LettersDocument5 pagesBioorganic & Medicinal Chemistry LettersDeden IndraDinataNo ratings yet
- La Densidad Del Potasio Que Tiene Una Estructura BCC Es 0.855 g/cm3 y Su Peso Atómico Es 39.09 G/mol. Calcular El Parámetro ReticularDocument4 pagesLa Densidad Del Potasio Que Tiene Una Estructura BCC Es 0.855 g/cm3 y Su Peso Atómico Es 39.09 G/mol. Calcular El Parámetro ReticularMarena Molano MendozaNo ratings yet
- 1 s2.0 S1658365514000831 MainDocument16 pages1 s2.0 S1658365514000831 Maintaoufik akabliNo ratings yet
- (Morning) : Favorskii Rearrangement and Its Synthetic ApplicationsDocument7 pages(Morning) : Favorskii Rearrangement and Its Synthetic ApplicationsMUhammad AsifNo ratings yet
- TEST 2 GOC & POC Tough by S.K.sinha See Chemistry Animations atDocument3 pagesTEST 2 GOC & POC Tough by S.K.sinha See Chemistry Animations atmyiitchemistryNo ratings yet
- Đư NGDocument4 pagesĐư NGPhong Chu VanNo ratings yet
- Article A New Carbazole-Based Helically Chiral Architecture: Synthesis and Physical PropertiesDocument4 pagesArticle A New Carbazole-Based Helically Chiral Architecture: Synthesis and Physical PropertiesMourad Ben BraiekNo ratings yet
- Chemistry Clinic ClassDocument5 pagesChemistry Clinic ClassWebby ZimbaNo ratings yet
- The Construction of High-Nuclearity Isopolyoxoniobates With Pentagonal Building Blocks: (HNB O) and (H NB O (Co) )Document4 pagesThe Construction of High-Nuclearity Isopolyoxoniobates With Pentagonal Building Blocks: (HNB O) and (H NB O (Co) )Libre Joel IanNo ratings yet
- Bai 2016Document7 pagesBai 2016Giang Hoang HuongNo ratings yet
- Symmetry 14 02496Document7 pagesSymmetry 14 02496Dipro SalicNo ratings yet
- ZN (II) Formate Bpy Coordination Polymer 2016Document5 pagesZN (II) Formate Bpy Coordination Polymer 2016Legeek GamingNo ratings yet
- SummativeTest-Q2 Gr. 9Document5 pagesSummativeTest-Q2 Gr. 9Chee MaRieNo ratings yet
- Flexible All-Perovskite Tandem Solar Cells Approaching 25% Efficiency With Molecule-Bridged Hole-Selective ContactDocument12 pagesFlexible All-Perovskite Tandem Solar Cells Approaching 25% Efficiency With Molecule-Bridged Hole-Selective ContactVeena SnaikNo ratings yet
- Ilovepdf MergedDocument215 pagesIlovepdf Merged亗『13 A M I V I 』亗No ratings yet
- Ans Chapter1 PDFDocument3 pagesAns Chapter1 PDFberyl gNo ratings yet
- CHP 3 - Atomic Structure & The Periodic Table 1 QPDocument8 pagesCHP 3 - Atomic Structure & The Periodic Table 1 QPDhrumeelNo ratings yet
- Jain 2013Document13 pagesJain 2013zahoorNo ratings yet
- Investigation of Polymer Mesophases by Optical MicrosDocument6 pagesInvestigation of Polymer Mesophases by Optical Microssoheil farshbafNo ratings yet
- 10.1038@s41929 018 0169 3Document8 pages10.1038@s41929 018 0169 3Patrik ConkaNo ratings yet
- Aldehyde, Ketone and Carboxylic acidPYQsJEEMainsDocument45 pagesAldehyde, Ketone and Carboxylic acidPYQsJEEMainsmjonfire3023No ratings yet
- L1 Measurement-StudentDocument66 pagesL1 Measurement-StudentNN JKNo ratings yet
- Síntese Na Superfície de Um Alótropo de Carbono Duplamente Anti-AromáticoDocument6 pagesSíntese Na Superfície de Um Alótropo de Carbono Duplamente Anti-AromáticoTony PereiraNo ratings yet
- Challenges and Outlook For Catalytic Direct Amidation ReactionsDocument5 pagesChallenges and Outlook For Catalytic Direct Amidation ReactionsAngélica Andrea SalinasNo ratings yet
- HydrocarbonsDocument131 pagesHydrocarbonsyeet LmaoNo ratings yet
- Kelompok 9Document7 pagesKelompok 9Muhammad Haksan PratamaNo ratings yet
- Synthesis, Enantiomeric Resolution and Optical Properties of 8-CyanohexaheliceneDocument4 pagesSynthesis, Enantiomeric Resolution and Optical Properties of 8-CyanohexaheliceneMourad Ben BraiekNo ratings yet
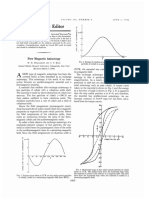
- New Magnetic Anisotropy13Document2 pagesNew Magnetic Anisotropy13安王No ratings yet
- High Resolution Nuclear Magnetic Resonance Spectroscopy: Volume 2From EverandHigh Resolution Nuclear Magnetic Resonance Spectroscopy: Volume 2No ratings yet
- The Principles of Heterocyclic ChemistryFrom EverandThe Principles of Heterocyclic ChemistryRating: 3 out of 5 stars3/5 (2)
- Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryFrom EverandOrganometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryF. G. A. StoneNo ratings yet
- XXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973From EverandXXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973No ratings yet
- Govt Projects 3Document48 pagesGovt Projects 3A BNo ratings yet
- Telephone Business Communication 1Document27 pagesTelephone Business Communication 1John Mark Agustin GarciaNo ratings yet
- Fea Stress Analysis of Drill BitDocument40 pagesFea Stress Analysis of Drill BitjagadeeshNo ratings yet
- Miracall PABX Telephone System PDFDocument1 pageMiracall PABX Telephone System PDFsundar chapagainNo ratings yet
- Electrostatics Module-IDocument8 pagesElectrostatics Module-IVarjula BalakrishnaNo ratings yet
- Reference Guide - Machine Vision - English A4Document40 pagesReference Guide - Machine Vision - English A4Geoff EricksonNo ratings yet
- الفطرياتDocument82 pagesالفطرياتKhaled AbdelfattahNo ratings yet
- How To Transfer Transactions To GLDocument5 pagesHow To Transfer Transactions To GLGuillermo ToddNo ratings yet
- ParamnesiaDocument3 pagesParamnesiaThe LullabyNo ratings yet
- List of TOOLS, EQUIPMENT AND MATERIALS Food ProcessingDocument9 pagesList of TOOLS, EQUIPMENT AND MATERIALS Food ProcessingCzar InaNo ratings yet
- Amcrps Az26-700nDocument2 pagesAmcrps Az26-700nLázaro MagalhãesNo ratings yet
- Mastering English - Form 2Document92 pagesMastering English - Form 2corine da83% (6)
- Soc 2230 SyllabusDocument8 pagesSoc 2230 SyllabusAnastasia IndrieNo ratings yet
- BS en 00931-1998Document20 pagesBS en 00931-1998Amer AmeryNo ratings yet
- Q2-Learning Plan in TLE-7Document3 pagesQ2-Learning Plan in TLE-7Lavinia PeterosNo ratings yet
- Assignment 1 - PROC 8170 - ENGI 91161Document2 pagesAssignment 1 - PROC 8170 - ENGI 91161latifmeijidaNo ratings yet
- The Revised MSX Red BookDocument302 pagesThe Revised MSX Red BookfreeTerp100% (1)
- Graph Theory Approach To Transportation Systems DesignDocument8 pagesGraph Theory Approach To Transportation Systems Designsandi nurmansyahNo ratings yet
- Stc750 Truck Crane 75 Tons Lifting Capacity: Quality Changes The WorldDocument9 pagesStc750 Truck Crane 75 Tons Lifting Capacity: Quality Changes The WorldDidier FernandezNo ratings yet
- MSTeams DiagnosticsDocument270 pagesMSTeams Diagnosticssafae daliNo ratings yet
- Rereading Rudyard Kipling's Kim in The Light of The Notion 'Dialogue of Civilizations': A Multicultural PerspectiveDocument14 pagesRereading Rudyard Kipling's Kim in The Light of The Notion 'Dialogue of Civilizations': A Multicultural Perspectivesakura yuimikoNo ratings yet
- Six Key Points To Merge Project Marketing Into Project ManagementDocument6 pagesSix Key Points To Merge Project Marketing Into Project Managementapi-3707091No ratings yet
- Manual of NPN - To - PNP ConverterDocument2 pagesManual of NPN - To - PNP ConverterLaboratório de Ensaios de MotoresNo ratings yet
- Habeas Corpus in International LawDocument17 pagesHabeas Corpus in International LawSalma Salsabila PutriningrumNo ratings yet
- Audit CIS 1.4Document4 pagesAudit CIS 1.4Patrick AlvinNo ratings yet
- Intervention PlanDocument5 pagesIntervention PlanShiela Marie OcampoNo ratings yet
- Daleys Notebook The Magic Memories 105Document8 pagesDaleys Notebook The Magic Memories 105AndreaClementePancotti100% (1)
- Element or CompoundDocument48 pagesElement or CompoundGLAIZA CALVARIONo ratings yet
- Impact of Artificial Intelligence On The Sports IndustryDocument6 pagesImpact of Artificial Intelligence On The Sports IndustryAshwin AsthanaNo ratings yet
- Peng Et Al. (2019)Document6 pagesPeng Et Al. (2019)Natasha MaharaniNo ratings yet