You might also like
- Electrochemical and Electrocatalytic Reactions of Carbon DioxideFrom EverandElectrochemical and Electrocatalytic Reactions of Carbon DioxideB.P. SullivanRating: 5 out of 5 stars5/5 (1)
- ChemCatChem - 2012 - Yu - The Unique Role of CaO in Stabilizing The PT Al2O3 Catalyst For The Dehydrogenation ofDocument6 pagesChemCatChem - 2012 - Yu - The Unique Role of CaO in Stabilizing The PT Al2O3 Catalyst For The Dehydrogenation ofSangHao NgNo ratings yet
- Article EvaluationOfCarbonPartitioningDocument16 pagesArticle EvaluationOfCarbonPartitioningEnriqe PuentesNo ratings yet
- High-Performance Nitrogen-Doped Intermetallic PtNi Catalyst For The Oxygen Reduction ReactionDocument30 pagesHigh-Performance Nitrogen-Doped Intermetallic PtNi Catalyst For The Oxygen Reduction ReactionSuganthi GaneshNo ratings yet
- ACS Catalysis 2020 Revealing The Role of Surface Composition On TheDocument23 pagesACS Catalysis 2020 Revealing The Role of Surface Composition On TheTanveer KhanNo ratings yet
- Lu 2021 PT - CeO2Document15 pagesLu 2021 PT - CeO2Raian Yousuf TanmoyNo ratings yet
- Combustion and Flame: J.A. Federici, D.G. VlachosDocument4 pagesCombustion and Flame: J.A. Federici, D.G. Vlachosalex royNo ratings yet
- 4.applied Catalysis BDocument8 pages4.applied Catalysis BCristianAtanasiuNo ratings yet
- Zou 2021Document7 pagesZou 202112334sohakamNo ratings yet
- Anie 202002394Document8 pagesAnie 202002394Ning ZhangNo ratings yet
- Engineering Pt-Mn2O3 Interface To Boost Selective Oxidation of Ethylene Glycol To Glycolic AcidDocument11 pagesEngineering Pt-Mn2O3 Interface To Boost Selective Oxidation of Ethylene Glycol To Glycolic Acid63011373No ratings yet
- JPCC 2008 112 1115 UVvis Xanes GoldDocument9 pagesJPCC 2008 112 1115 UVvis Xanes GoldUser7785No ratings yet
- HhjoDocument14 pagesHhjoHarshitha Maria RockNo ratings yet
- Studies of The Influence of Alloying Elements On T 2 PDFDocument7 pagesStudies of The Influence of Alloying Elements On T 2 PDFSonu Kumar MishraNo ratings yet
- Atomistic Modeling of Carbon Cottrell Atmospheres in BCC IronDocument9 pagesAtomistic Modeling of Carbon Cottrell Atmospheres in BCC Ironle hoangNo ratings yet
- Synthesis-Structure-Activity Relationships in Co O Catalyzed CO OxidationDocument12 pagesSynthesis-Structure-Activity Relationships in Co O Catalyzed CO OxidationmanuelNo ratings yet
- 1 s2.0 S0169433221023795 MainDocument11 pages1 s2.0 S0169433221023795 MainDaniel MontalvoNo ratings yet
- 1 s2.0 S0926337323007622 MainDocument12 pages1 s2.0 S0926337323007622 Mainzhizheng wuNo ratings yet
- Catalytic Synthesis of Methanol From CO/H,: Journ.4L of C 4talysisDocument23 pagesCatalytic Synthesis of Methanol From CO/H,: Journ.4L of C 4talysisulsakNo ratings yet
- Catalysis Today: Oscar G Omez C Apiro, Karen A. Aravena Riquelme, Romel Jim Enez, Luis E. Arteaga-P Erez, PHDDocument7 pagesCatalysis Today: Oscar G Omez C Apiro, Karen A. Aravena Riquelme, Romel Jim Enez, Luis E. Arteaga-P Erez, PHDEcNo ratings yet
- 2013 CJCEJinDocument8 pages2013 CJCEJinNadineNo ratings yet
- Adsorption and Desorption of Carbon Dioxide and Nitrogen On Zeolite 5A PDFDocument19 pagesAdsorption and Desorption of Carbon Dioxide and Nitrogen On Zeolite 5A PDFBình Tân LêNo ratings yet
- Accepted Manuscript: Accepted Manuscripts Are Published Online Shortly AfterDocument10 pagesAccepted Manuscript: Accepted Manuscripts Are Published Online Shortly AfterDada GiltronNo ratings yet
- Aranzabal 2009Document9 pagesAranzabal 2009bruno barrosNo ratings yet
- Gwak (2020) - Analyzing Oxygen Transport ResistanceDocument14 pagesGwak (2020) - Analyzing Oxygen Transport ResistanceGeonhui GwakNo ratings yet
- Bor 2Document8 pagesBor 2petru apopeiNo ratings yet
- Studiu Fullerene U.S. Army ResearchDocument37 pagesStudiu Fullerene U.S. Army ResearchTudor RatiuNo ratings yet
- Applied Surface Science: Yan Wang, Xilin Zhang, Cheng Cheng, Zongxian YangDocument7 pagesApplied Surface Science: Yan Wang, Xilin Zhang, Cheng Cheng, Zongxian YangHóa PinkyNo ratings yet
- Low-Temperature Hydrogenation of The C O Bond of Propanal Over Ni-Pt Bimetallic Catalysts: From Model Surfaces To Supported CatalystsDocument7 pagesLow-Temperature Hydrogenation of The C O Bond of Propanal Over Ni-Pt Bimetallic Catalysts: From Model Surfaces To Supported CatalystsAmir DanialNo ratings yet
- Xia Et Al-Angewandte Chemie International EditionDocument29 pagesXia Et Al-Angewandte Chemie International EditionKatiane MesquitaNo ratings yet
- Effect of Electrode PT Loading and Cathode Flow Field Plate Type On The Degradation of Pemfc Qu Full ChapterDocument31 pagesEffect of Electrode PT Loading and Cathode Flow Field Plate Type On The Degradation of Pemfc Qu Full Chapterdale.rolling783100% (4)
- Single-Atom Catalysis of Co Oxidation Using PT 1 /feo X: Nature Chemistry July 2011Document9 pagesSingle-Atom Catalysis of Co Oxidation Using PT 1 /feo X: Nature Chemistry July 2011Nina TaraNo ratings yet
- 2005 Kamisaka - Theoretical Study of The Structure and Optical Properties ofDocument10 pages2005 Kamisaka - Theoretical Study of The Structure and Optical Properties ofHeri RustamajiNo ratings yet
- CoCeO2 and NiCeO2 Catalysts For Ethanol Steam Reforming Effect of TheDocument20 pagesCoCeO2 and NiCeO2 Catalysts For Ethanol Steam Reforming Effect of TheDana MateiNo ratings yet
- Single-Atom Catalysis Using Pt/Graphene Achieved Through Atomic Layer DepositionDocument10 pagesSingle-Atom Catalysis Using Pt/Graphene Achieved Through Atomic Layer DepositionMogan RajNo ratings yet
- Wang 2017Document31 pagesWang 2017valentinaNo ratings yet
- Energies 16 03659Document15 pagesEnergies 16 03659AlisaNo ratings yet
- 1 AntoniusDocument6 pages1 Antoniusapi-3728640No ratings yet
- Hrtem PTCDocument6 pagesHrtem PTCLuis Ramón GómezNo ratings yet
- Electronic Structure Lattice Energies and Born ExpDocument10 pagesElectronic Structure Lattice Energies and Born ExpAdolfNo ratings yet
- Microkinetic Analysis of Acetone Hydrogenation Over Pt-SiO2Document16 pagesMicrokinetic Analysis of Acetone Hydrogenation Over Pt-SiO2李国俊No ratings yet
- Accepted Manuscript: Catalysis TodayDocument25 pagesAccepted Manuscript: Catalysis TodayGonzalo BenavidesNo ratings yet
- Zhu 2015Document10 pagesZhu 2015reclatis14No ratings yet
- Temperature-Programmed Oxidation of Coked Noble Metal Catalysts After Autothermal Reforming of N-HexadecaneDocument9 pagesTemperature-Programmed Oxidation of Coked Noble Metal Catalysts After Autothermal Reforming of N-HexadecaneImran KhanNo ratings yet
- Catalyst Surface Modification by Ion-Beam Sputtering: Resume of Dr. Nancy Artioli LectureDocument5 pagesCatalyst Surface Modification by Ion-Beam Sputtering: Resume of Dr. Nancy Artioli LectureDanie Moreno DamanikNo ratings yet
- 10 1021@acs JPCC 0c04594Document38 pages10 1021@acs JPCC 0c04594rajanadarajanNo ratings yet
- Cellulose Ag3PO4Document6 pagesCellulose Ag3PO4Izelgue ChouaaibNo ratings yet
- Perbandingan Antara Protokol NIOSH, IMPROVE - A, Dan EUSAAR - 2 Menemukan Protokol Termal-Optik Yang Optimal Untuk Sampel OCEC FilipinaDocument9 pagesPerbandingan Antara Protokol NIOSH, IMPROVE - A, Dan EUSAAR - 2 Menemukan Protokol Termal-Optik Yang Optimal Untuk Sampel OCEC FilipinaharyawatiNo ratings yet
- Palladium Catalysts On Activated Carbon Supports IDocument16 pagesPalladium Catalysts On Activated Carbon Supports IScott PilgrimNo ratings yet
- Angew Chem Int Ed - 2014 - Filot - The Optimally Performing Fischer Tropsch CatalystDocument5 pagesAngew Chem Int Ed - 2014 - Filot - The Optimally Performing Fischer Tropsch CatalystTrinityRVxNo ratings yet
- Acscatal 8b03765Document13 pagesAcscatal 8b03765Yu YanNo ratings yet
- Angew Chem Int Ed - 2023 - Li - Air Stable Organoradical Boron ReagentsDocument7 pagesAngew Chem Int Ed - 2023 - Li - Air Stable Organoradical Boron ReagentsTutu CaiNo ratings yet
- Jiang 2015Document44 pagesJiang 2015Aisah Cory PrasonoNo ratings yet
- 1 OnlineDocument17 pages1 Online1592162022No ratings yet
- Co2 Activation Over Catalytic Surfaces: Chemphyschem August 2017Document10 pagesCo2 Activation Over Catalytic Surfaces: Chemphyschem August 2017sheraz ahmadNo ratings yet
- Crystals 13 00255 v2Document13 pagesCrystals 13 00255 v2Menjamin SalasNo ratings yet
- Polymer: ArticleinfoDocument9 pagesPolymer: ArticleinfoDelcys PazNo ratings yet
- Kinetics of The Fischer Tropsch Reaction in Fixed Bed Reactor Over A Nano Structured Fe Co Ce Catalyst Supported With SiO2Document9 pagesKinetics of The Fischer Tropsch Reaction in Fixed Bed Reactor Over A Nano Structured Fe Co Ce Catalyst Supported With SiO2Pfunzo PharmelaNo ratings yet
- Ultrasound-Triggered Sonocatalytic Reduction of CO2 Via H2Ti3O7 NanowiresDocument7 pagesUltrasound-Triggered Sonocatalytic Reduction of CO2 Via H2Ti3O7 NanowiresPrince SharmaNo ratings yet
- Catalytic Ammonia Oxidation On Platinum: Mechanism and Catalyst Restructuring at High and Low PressureDocument20 pagesCatalytic Ammonia Oxidation On Platinum: Mechanism and Catalyst Restructuring at High and Low PressureNabil RadianNo ratings yet
- Kinetic Assessment of The Simultaneous Hydrodesulfurization of Dibenzothiophene and The Hydrogenation of Diverse Polyaromatic Structures (2018)Document17 pagesKinetic Assessment of The Simultaneous Hydrodesulfurization of Dibenzothiophene and The Hydrogenation of Diverse Polyaromatic Structures (2018)marcos carrilloNo ratings yet
- The Functionalities of PT - Al2O3 CatalyDocument11 pagesThe Functionalities of PT - Al2O3 Catalymarcos carrilloNo ratings yet
- Dorneles de Mello 2018Document9 pagesDorneles de Mello 2018marcos carrilloNo ratings yet
- Hydrodesulfurization of Dibenzothiophene in A Micro Trickle Bed Catalytic Reactor Under Operating Conditions From ReactiveDocument15 pagesHydrodesulfurization of Dibenzothiophene in A Micro Trickle Bed Catalytic Reactor Under Operating Conditions From Reactivemarcos carrilloNo ratings yet
- Journal of Catalysis: Xuerong Zhou, Xiang Li, Roel Prins, Anjie Wang, Lin Wang, Shengnan Liu, Qiang ShengDocument12 pagesJournal of Catalysis: Xuerong Zhou, Xiang Li, Roel Prins, Anjie Wang, Lin Wang, Shengnan Liu, Qiang Shengmarcos carrilloNo ratings yet
- Experimental Methods in Catalytic KineticsDocument13 pagesExperimental Methods in Catalytic Kineticsmarcos carrilloNo ratings yet
- 4356 - Problems and ChalengesDocument53 pages4356 - Problems and Chalengesdownload100% (1)
- Catalysis Reviews: Science and EngineeringDocument45 pagesCatalysis Reviews: Science and Engineeringmarcos carrilloNo ratings yet
- Corrosion Control in IndustryDocument36 pagesCorrosion Control in Industrymarcos carrilloNo ratings yet
- Kalpakka Chemical Cems Process Diagram-Model - PDF NewDocument1 pageKalpakka Chemical Cems Process Diagram-Model - PDF NewRajesh KumarNo ratings yet
- Making Herbal Baths Salts: Presented By: Di-Di HoffmanDocument12 pagesMaking Herbal Baths Salts: Presented By: Di-Di HoffmanGamal Al IsraNo ratings yet
- Chemistry Powerpoint (Autosaved)Document57 pagesChemistry Powerpoint (Autosaved)Zeedan MohammedNo ratings yet
- Cast It2000Document5 pagesCast It2000jkpolino85No ratings yet
- Welding of P91Document37 pagesWelding of P91nishant361100% (4)
- Microbiology and Molecular Biology Reviews-1994-Robinow-211.fullDocument22 pagesMicrobiology and Molecular Biology Reviews-1994-Robinow-211.fullBruno RivoiraNo ratings yet
- ABI NapoleonDocument16 pagesABI NapoleonFachri RoziNo ratings yet
- Plasma Arc Welding 19d41a0329 FinalDocument36 pagesPlasma Arc Welding 19d41a0329 Final20-119 KATRAVATH HARINo ratings yet
- Mixtures Solutions & Mechanical Mixtures'-2Document17 pagesMixtures Solutions & Mechanical Mixtures'-2Mariam AmgadNo ratings yet
- Safety Data Sheet: Section 1. IdentificationDocument10 pagesSafety Data Sheet: Section 1. IdentificationFredy Fernando FuentesNo ratings yet
- Nomenclature Worksheet Part 1Document8 pagesNomenclature Worksheet Part 1Jocelyn MarmolNo ratings yet
- w328 Concentration Worksheet PDFDocument2 pagesw328 Concentration Worksheet PDFtheresia gallaNo ratings yet
- Piusi Air Operated P60 1 InstructionsDocument2 pagesPiusi Air Operated P60 1 Instructions673348300 Producto CartesNo ratings yet
- Topic 1 Manual 4th Edition Ver1 2023Document44 pagesTopic 1 Manual 4th Edition Ver1 2023Ha NaNo ratings yet
- Jsa PVC, CPVC, Teflon, PVDFDocument7 pagesJsa PVC, CPVC, Teflon, PVDFM. MagnoNo ratings yet
- Manufacturing of Cement 2023. CivilDocument43 pagesManufacturing of Cement 2023. CivilRoony RoyNo ratings yet
- NCERT Table Important TrendsDocument18 pagesNCERT Table Important TrendsDeep ZalkeNo ratings yet
- Astm F1941M 2007Document12 pagesAstm F1941M 2007Jesse ChenNo ratings yet
- Amines Ii-1Document25 pagesAmines Ii-1Abhishek PatilNo ratings yet
- MercerizationDocument18 pagesMercerizationFarabbi AdittoNo ratings yet
- IER - Normal RegenerationDocument2 pagesIER - Normal RegenerationmuraliNo ratings yet
- Ledm0126 01Document9 pagesLedm0126 01Djebali MouradNo ratings yet
- Learning Activity Sheet Science 6 Week 1 CompetenciesDocument8 pagesLearning Activity Sheet Science 6 Week 1 CompetenciesShekaina Faith LozadaNo ratings yet
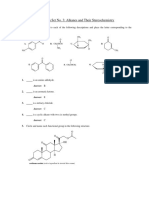
- Problem Set 3 - Alkanes and StereochemDocument6 pagesProblem Set 3 - Alkanes and StereochemKatrina Louise GonzalesNo ratings yet
- Booklet Sr. NoDocument16 pagesBooklet Sr. NoAnshul SoniNo ratings yet
- Candle LabDocument5 pagesCandle LabHarrison Lee50% (2)
- Recyllose - Products From Sewage: Turning Wastewater Into A Cellulose-MineDocument5 pagesRecyllose - Products From Sewage: Turning Wastewater Into A Cellulose-MineLalit Mohan Senapati100% (1)
- 08-12-2022Document26 pages08-12-2022Hendy Kilat BuanaNo ratings yet
- Vancomycin SolutionDocument1 pageVancomycin SolutionjamalNo ratings yet
- Astm Astm d2887Document20 pagesAstm Astm d2887Orlando Rojas100% (1)