You might also like
- Bio Placement Test Review Questions Review 1: Answer Key On Page 11 Select The Correct AnswerDocument31 pagesBio Placement Test Review Questions Review 1: Answer Key On Page 11 Select The Correct AnswerAndrea RaquilNo ratings yet
- Epigenetic Influences and DiseaseDocument69 pagesEpigenetic Influences and DiseasescribdremoNo ratings yet
- Cell Structure and Function PDFDocument15 pagesCell Structure and Function PDFAbdul RahmanNo ratings yet
- Biotechnology 8 Q2 Manipulation of Genetic MaterialDocument10 pagesBiotechnology 8 Q2 Manipulation of Genetic MaterialCold CoockiesNo ratings yet
- Fragged Citadel WIPDocument13 pagesFragged Citadel WIPCole Parker100% (2)
- Acta Oncol 2023 May 7 Jorgensen P LDocument8 pagesActa Oncol 2023 May 7 Jorgensen P LBALTAZAR OTTONELLONo ratings yet
- Steiner TDocument7 pagesSteiner TBabu MolinaNo ratings yet
- Gao 2015Document17 pagesGao 2015saherrodriguezNo ratings yet
- Epigenetics and MicroRNAs.5Document6 pagesEpigenetics and MicroRNAs.5Saurabh GayaliNo ratings yet
- Kopinski 2021Document15 pagesKopinski 2021lu veNo ratings yet
- Cancer Epigenetics: Peter W. LairdDocument12 pagesCancer Epigenetics: Peter W. LairdCarlos AlbertoNo ratings yet
- Molecules: Duchenne Muscular Dystrophy: From Diagnosis To TherapyDocument17 pagesMolecules: Duchenne Muscular Dystrophy: From Diagnosis To TherapyzzooooeeeeeeNo ratings yet
- IA - Molecular Biology of Pharmacology 160908Document39 pagesIA - Molecular Biology of Pharmacology 160908Susi RutmalemNo ratings yet
- Use of CisplatinDocument14 pagesUse of CisplatinsmijatovicibissNo ratings yet
- CisplatinDocument15 pagesCisplatinBrantas Pra AzariNo ratings yet
- Yokochi 2004Document6 pagesYokochi 2004hasanbatuhan küçükNo ratings yet
- Epigenetic Modulations and Lineage Plasticity in Advanced Prostate CancerDocument10 pagesEpigenetic Modulations and Lineage Plasticity in Advanced Prostate CancerGabriel ŞarguNo ratings yet
- Dusl 2015Document9 pagesDusl 2015tkd assassinNo ratings yet
- EZH2: Not EZHY (Easy) To Deal: ReviewDocument16 pagesEZH2: Not EZHY (Easy) To Deal: ReviewLorena RamosNo ratings yet
- Cancer UlDocument10 pagesCancer UlCelatuchiacNo ratings yet
- MEK Inhibitors Activate WNT Signalling and Induce Stem Cell Plasticity in Colorectal CancerDocument17 pagesMEK Inhibitors Activate WNT Signalling and Induce Stem Cell Plasticity in Colorectal CancerYuqing LiangNo ratings yet
- Connecting Chromatin Modifying Factors To DNA Damage ResponseDocument15 pagesConnecting Chromatin Modifying Factors To DNA Damage Responsedjum4liNo ratings yet
- Giacci A 2016Document9 pagesGiacci A 2016yagami19lightNo ratings yet
- Dna Methylation DissertationDocument6 pagesDna Methylation DissertationCustomWrittenPaperLittleRock100% (1)
- (B) On (E) - Cohistones and The Epigenetic Alterations at The Root of Bone CancerDocument12 pages(B) On (E) - Cohistones and The Epigenetic Alterations at The Root of Bone CancerJudeNo ratings yet
- Matrix Metalloproteinases/Tissue Inhibitors of MetalloproteinasesDocument9 pagesMatrix Metalloproteinases/Tissue Inhibitors of MetalloproteinasesireneaureliaNo ratings yet
- Clinical Genetics - 2011 - Kanwal - Epigenetic Modifications in CancerDocument9 pagesClinical Genetics - 2011 - Kanwal - Epigenetic Modifications in Cancerpharm.ajbashirNo ratings yet
- NIH Public Access: Author ManuscriptDocument20 pagesNIH Public Access: Author Manuscriptfranciscrick69No ratings yet
- Bnab 014Document38 pagesBnab 014RodrigoNo ratings yet
- Mitochondrial DNA Mutations in Human DiseaseDocument9 pagesMitochondrial DNA Mutations in Human DiseaseLuna SmithNo ratings yet
- Post Translational Modifications On B Myosin Heavy Chain 1653708664Document24 pagesPost Translational Modifications On B Myosin Heavy Chain 1653708664Happy InsightsNo ratings yet
- 2000-Drug Development ResearchDocument10 pages2000-Drug Development ResearchEmerson CasaliNo ratings yet
- JAMA Leukemia 1Document3 pagesJAMA Leukemia 1indra_gosalNo ratings yet
- FTPDocument7 pagesFTPAlema PelesićNo ratings yet
- Mitochondrial DDocument17 pagesMitochondrial DIt's MeNo ratings yet
- 483 Final HomeworkDocument6 pages483 Final HomeworkzeynoleeeNo ratings yet
- Groen 2018Document12 pagesGroen 2018Felipe MNo ratings yet
- 5-Azacitidine in Aggressive Myelodysplastic Syndromes Regulates Chromatin Structure at PU.1 Gene and Cell Differentiation CapacityDocument8 pages5-Azacitidine in Aggressive Myelodysplastic Syndromes Regulates Chromatin Structure at PU.1 Gene and Cell Differentiation CapacitycherruskaNo ratings yet
- Expresion Patterns of Genes, Regulating Lipid Metabolism in Prostate TumorsDocument16 pagesExpresion Patterns of Genes, Regulating Lipid Metabolism in Prostate TumorsАнна ШаповаловаNo ratings yet
- TET2 Decrease - in - Lymphoid - Specific - Helicase - and - 5-HydroDocument13 pagesTET2 Decrease - in - Lymphoid - Specific - Helicase - and - 5-Hydromejia_jpNo ratings yet
- 225.full, Efek Pemberian Statin Rendah Terhadap LipidDocument24 pages225.full, Efek Pemberian Statin Rendah Terhadap Lipidetik ainun rohmahNo ratings yet
- Arzuman 2014 BDocument14 pagesArzuman 2014 BMonikaNo ratings yet
- Can 13 0527rDocument30 pagesCan 13 0527rData KiswaraNo ratings yet
- Tugas Bu AniDocument23 pagesTugas Bu AniYon-SyuhandaNo ratings yet
- Dahl-Guldberg2003 Article DNAMethylationAnalysisTechniquDocument18 pagesDahl-Guldberg2003 Article DNAMethylationAnalysisTechniquVanessa MillarNo ratings yet
- Site-To-Site Interdomain Communication May Mediate Different Loss-Of-Function Mechanisms in A Cancer-Associated NQO1 PolymorphismDocument18 pagesSite-To-Site Interdomain Communication May Mediate Different Loss-Of-Function Mechanisms in A Cancer-Associated NQO1 PolymorphismDavid Galiano LatorreNo ratings yet
- PHD Thesis Dna MethylationDocument4 pagesPHD Thesis Dna MethylationEmily Smith100% (2)
- Epigenetic and Cancer Therapy, 2019Document12 pagesEpigenetic and Cancer Therapy, 2019AbsjeyNo ratings yet
- Structure, Mechanism, and Regulation of Polycomb-Repressive Complex 2Document10 pagesStructure, Mechanism, and Regulation of Polycomb-Repressive Complex 2sofiaNo ratings yet
- Targeting Neddylation Pathway With MLN4924 (Pevonedistat) InducesDocument6 pagesTargeting Neddylation Pathway With MLN4924 (Pevonedistat) InducescarlakerengrNo ratings yet
- BAI Et Al-2004-Annals of The New York Academy of SciencesDocument6 pagesBAI Et Al-2004-Annals of The New York Academy of SciencesAulas EspañolNo ratings yet
- Hao 2012Document9 pagesHao 2012vickydivi09No ratings yet
- Metabolites: Comparative Untargeted Metabolomic Profiling of Induced Mitochondrial Fusion in Pancreatic CancerDocument22 pagesMetabolites: Comparative Untargeted Metabolomic Profiling of Induced Mitochondrial Fusion in Pancreatic CancerSukma's Wahyulii TiyasNo ratings yet
- Review: Epigenetics in Human Obesity and Type 2 DiabetesDocument17 pagesReview: Epigenetics in Human Obesity and Type 2 DiabetesStephany BoninNo ratings yet
- Epigenetics in CancerDocument10 pagesEpigenetics in CancermartinNo ratings yet
- 9 Sudemycins 2011Document8 pages9 Sudemycins 2011milenerato2240No ratings yet
- Inaba 2010Document11 pagesInaba 2010Eduardo GirónNo ratings yet
- Intersectin Regulates Dendritic Spine Development and Somatodendritic Endocytosis But Not Synaptic Vesicle Recycling in Hippocampal NeuronsDocument10 pagesIntersectin Regulates Dendritic Spine Development and Somatodendritic Endocytosis But Not Synaptic Vesicle Recycling in Hippocampal NeuronsSergeat18BNo ratings yet
- Biochemical and Molecular Analysis in A Patient With The Severe Form of Hunter Syndrome After Bone Marrow TransplantationDocument5 pagesBiochemical and Molecular Analysis in A Patient With The Severe Form of Hunter Syndrome After Bone Marrow TransplantationmgNo ratings yet
- High Expression of Oxidative Phosphorylation Genes Predicts Improved Survival in Squamous Cell Carcinomas of The Head and Neck and LungDocument14 pagesHigh Expression of Oxidative Phosphorylation Genes Predicts Improved Survival in Squamous Cell Carcinomas of The Head and Neck and Lungsalmaa safiraNo ratings yet
- Biomedicine & Pharmacotherapy: SciencedirectDocument13 pagesBiomedicine & Pharmacotherapy: SciencedirectAdrian BratuNo ratings yet
- Exercise Induces Muscle Fiber Type SwitchingDocument10 pagesExercise Induces Muscle Fiber Type SwitchingbookNo ratings yet
- VIP Cives Oncotarget 2016Document16 pagesVIP Cives Oncotarget 2016Jemma ArakelyanNo ratings yet
- Bi5b00969 Si 001Document4 pagesBi5b00969 Si 001Jon RumbleyNo ratings yet
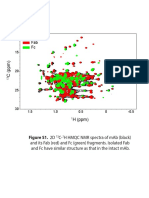
- Fab FC Mab: Figure S1. 2DDocument3 pagesFab FC Mab: Figure S1. 2DJon RumbleyNo ratings yet
- Bi6b01117 Si 001Document1 pageBi6b01117 Si 001Jon RumbleyNo ratings yet
- M-Value M-Value: 0 Unf M 0 Unf MDocument6 pagesM-Value M-Value: 0 Unf M 0 Unf MJon RumbleyNo ratings yet
- Acs Omega 2020Document17 pagesAcs Omega 2020Jon RumbleyNo ratings yet
- Biophysical Analysis of The Effect of Chemical Modification by 4-Oxononenal On The Structure, Stability, and Function of Binding Immunoglobulin Protein (BiP)Document21 pagesBiophysical Analysis of The Effect of Chemical Modification by 4-Oxononenal On The Structure, Stability, and Function of Binding Immunoglobulin Protein (BiP)Jon RumbleyNo ratings yet
- Cot CurveDocument16 pagesCot CurveVidyasagar Deshpande100% (1)
- Lesson 1 Cells The Building Blocks of LifeDocument15 pagesLesson 1 Cells The Building Blocks of LifeGwen MarielleNo ratings yet
- Ebook PDF Statistics Principles and Methods 8th Edition PDFDocument41 pagesEbook PDF Statistics Principles and Methods 8th Edition PDFjoseph.corey248100% (33)
- In Memory of Professor Biao Ding 1960 - 2015Document7 pagesIn Memory of Professor Biao Ding 1960 - 2015Balasubramanian PonnuswamiNo ratings yet
- Biotic Community ConceptDocument2 pagesBiotic Community Conceptmichaeluriel100% (2)
- Guardant360 A0771104 v1 FinalDocument6 pagesGuardant360 A0771104 v1 FinaltraveltreatsteluguNo ratings yet
- LAB 21 - Evolution and ClassificationDocument13 pagesLAB 21 - Evolution and ClassificationMary Vienne PascualNo ratings yet
- AQA Biology 8 End-Of-Chapter Answers PDFDocument2 pagesAQA Biology 8 End-Of-Chapter Answers PDFAparupa Radhika Devi DasiNo ratings yet
- Resent Status of Newborn Screening (NBS) in Bangladesh: Health Professionals' PerspectiveDocument23 pagesResent Status of Newborn Screening (NBS) in Bangladesh: Health Professionals' PerspectiveRahatul IslamNo ratings yet
- Polymerase Chain Reaction (PCR) : Key PointsDocument17 pagesPolymerase Chain Reaction (PCR) : Key PointsMaryem SafdarNo ratings yet
- Alelo Dominante Alelo RecesivoDocument3 pagesAlelo Dominante Alelo RecesivoValeria DelgadilloNo ratings yet
- Madison Grant, Maxwell Perkins, and Eugenics PublishingDocument27 pagesMadison Grant, Maxwell Perkins, and Eugenics PublishinglbburgessNo ratings yet
- TranslationDocument7 pagesTranslationFathimaNo ratings yet
- 1ST Exam 9-10Document8 pages1ST Exam 9-10Joanne GodezanoNo ratings yet
- Yearbook 2023Document395 pagesYearbook 2023riyazsalva90No ratings yet
- Flashcards - Topic 7.2 Factors Affecting Gene Expression - Edexcel (B) Biology A-LevelDocument19 pagesFlashcards - Topic 7.2 Factors Affecting Gene Expression - Edexcel (B) Biology A-LevelPrasanthanNo ratings yet
- Lesson 5 Hormones Heredity and EnvironmentDocument2 pagesLesson 5 Hormones Heredity and EnvironmentDony CaveNo ratings yet
- Rubric Protein Synthesis Flow ChartDocument1 pageRubric Protein Synthesis Flow ChartJohn OsborneNo ratings yet
- Chapter 2: (Genetics) : Heredity, Inheritance and VariationDocument11 pagesChapter 2: (Genetics) : Heredity, Inheritance and VariationRonan BartolazoNo ratings yet
- סיכום שיעורים 1-6 נוירוביולוגיהDocument66 pagesסיכום שיעורים 1-6 נוירוביולוגיהNika KielNo ratings yet
- Topic 1.2 Skeleton NotesDocument14 pagesTopic 1.2 Skeleton NotesNicholasNo ratings yet
- Enterococcus Faecium 129 BIO 3B Is Classified As Enterococcus Lactis 129 BIO 3BDocument1 pageEnterococcus Faecium 129 BIO 3B Is Classified As Enterococcus Lactis 129 BIO 3BJake FinnNo ratings yet
- Prokaryotic and Eukaryotic CellsDocument9 pagesProkaryotic and Eukaryotic CellsKwo Chai Wing100% (1)
- LM Science 11 1.11Document17 pagesLM Science 11 1.11panomo nasabyNo ratings yet
- 5d83b684c0fa4b974de9b431 - HS Biology 1 - Course #2000310Document7 pages5d83b684c0fa4b974de9b431 - HS Biology 1 - Course #2000310Lerante LaubaxNo ratings yet
- A Review of Methods Studying Diversity Soils ': Microbial inDocument7 pagesA Review of Methods Studying Diversity Soils ': Microbial inNancy HarletNo ratings yet