You might also like
- Test Bank For Microbiology A Systems Approach 6th Edition Marjorie Kelly Cowan Heidi SmithDocument31 pagesTest Bank For Microbiology A Systems Approach 6th Edition Marjorie Kelly Cowan Heidi SmithAmandaReynoldsagfcy100% (23)
- Peptidomimetics FINAL PDFDocument21 pagesPeptidomimetics FINAL PDFVissarapu NagalakshmiNo ratings yet
- In Silico Screening of Chalcone Derivatives As Promising EGFR TKDocument15 pagesIn Silico Screening of Chalcone Derivatives As Promising EGFR TKHalimi Fajri YazdanyarNo ratings yet
- Art:10.1007/s00044 014 1114 XDocument19 pagesArt:10.1007/s00044 014 1114 Xسعيد راضيNo ratings yet
- Siddiqui 2013Document7 pagesSiddiqui 2013regis.farias95No ratings yet
- địa liền inflammatory PDFDocument8 pagesđịa liền inflammatory PDFTâm PhanNo ratings yet
- Synthesis of Novel N-Substitutedphenyl-6-Oxo-3-Phenylpyridazine Derivatives As Cyclooxygenase-2 InhibitorsDocument12 pagesSynthesis of Novel N-Substitutedphenyl-6-Oxo-3-Phenylpyridazine Derivatives As Cyclooxygenase-2 InhibitorsHamza AssilaNo ratings yet
- 1 s2.0 S0102695X16300217 MainDocument10 pages1 s2.0 S0102695X16300217 MainPutri RizkyNo ratings yet
- 1 Research PaperDocument11 pages1 Research PaperSyeda Farah ShahNo ratings yet
- 16 7 2019 IsolationDocument8 pages16 7 2019 IsolationNgan HoangNo ratings yet
- Sun 2015Document6 pagesSun 2015Wina WildayantiNo ratings yet
- GC Analiza 2Document7 pagesGC Analiza 2vlad valuNo ratings yet
- Synthesis of Some Novel C-5 and N-3 Substituted 2 - (2-Hydroxyphenylimino) Thiazolidin-4-One Derivatives With Broad-Spectrum Antimicrobial ActivityDocument15 pagesSynthesis of Some Novel C-5 and N-3 Substituted 2 - (2-Hydroxyphenylimino) Thiazolidin-4-One Derivatives With Broad-Spectrum Antimicrobial Activityamit8109No ratings yet
- Medicinal ChemistryDocument9 pagesMedicinal ChemistryANBU DINESHNo ratings yet
- Journal Fitokim 1Document7 pagesJournal Fitokim 1Wiwin LatifahNo ratings yet
- TC 06052Document7 pagesTC 06052Aiko Cheryl SalsabilaNo ratings yet
- 05 Intan Soraya Che Sulaiman - Paling FunctionDocument14 pages05 Intan Soraya Che Sulaiman - Paling FunctionIdham ZaharudieNo ratings yet
- Jhet 4331Document12 pagesJhet 4331Hida NurulNo ratings yet
- 1 s2.0 S0753332216328062 MainDocument8 pages1 s2.0 S0753332216328062 Mainmeeret zerihunNo ratings yet
- Bioassay-Guided Isolation of Sesquiterpene Coumarins From Ferula Narthex Bioss: A New Anticancer AgentDocument6 pagesBioassay-Guided Isolation of Sesquiterpene Coumarins From Ferula Narthex Bioss: A New Anticancer AgentHamdi PutraNo ratings yet
- Joshi2008 PDFDocument8 pagesJoshi2008 PDFNguyễn Đức Tri ThứcNo ratings yet
- 4 Pharmacophoric ModelingDocument12 pages4 Pharmacophoric ModelingDr. Meenakshi DhanawatNo ratings yet
- Nueva MoleculaDocument7 pagesNueva MoleculaChuy MartinezNo ratings yet
- Molecules 28 06521 v3Document32 pagesMolecules 28 06521 v3kabikant chaurasiyasNo ratings yet
- Design, Synthesis, and Biological Activity of A Novel Series of BenzofuranDocument7 pagesDesign, Synthesis, and Biological Activity of A Novel Series of BenzofuranMario Suarez GiraldoNo ratings yet
- Pirazolona Articulo para Ejercicio MecanismoDocument22 pagesPirazolona Articulo para Ejercicio MecanismoJosé Guadalupe García EstradaNo ratings yet
- Synthesis in Vitro Safety and Antioxidant Activity of New Pyrrole Hydrazones2020acta PharmaceuticaOpen AccessDocument22 pagesSynthesis in Vitro Safety and Antioxidant Activity of New Pyrrole Hydrazones2020acta PharmaceuticaOpen AccessJose BarajasNo ratings yet
- Thiazole PaperDocument9 pagesThiazole PaperPrateek TyagiNo ratings yet
- PlectranthusDocument8 pagesPlectranthusTAUFIK MUHAMMAD FAKIHNo ratings yet
- 10.1007-s00044-012-0343-0Document8 pages10.1007-s00044-012-0343-0virenderchem79No ratings yet
- Bioorganic ChemistryDocument15 pagesBioorganic ChemistryAhmed AmerNo ratings yet
- Synthesis and characterization of new triazole derivatives with antifungal activityDocument5 pagesSynthesis and characterization of new triazole derivatives with antifungal activityAlhassan AlgizyNo ratings yet
- 5 Prakasha+new+article PDFDocument6 pages5 Prakasha+new+article PDFiajpsNo ratings yet
- tmp3EF5 TMPDocument7 pagestmp3EF5 TMPFrontiersNo ratings yet
- Synthesis and Biological Evaluation of Pyrrolo (2,3-b) Pyridine Analogues PDFDocument12 pagesSynthesis and Biological Evaluation of Pyrrolo (2,3-b) Pyridine Analogues PDFMiguelAlejandroMantaChavezNo ratings yet
- Synthesis and Pharmacological Evaluation of Novel Coumarin DerivativesDocument11 pagesSynthesis and Pharmacological Evaluation of Novel Coumarin Derivativesمصطفى باسم هادي B1No ratings yet
- Artigo CientificoDocument8 pagesArtigo CientificoELISANGELA SILVANo ratings yet
- Bauhinia KockianaDocument9 pagesBauhinia Kockianaromanauli situmorangNo ratings yet
- Novel Hybrid Molecules of Isoxazole Chalcone Derivatives: Synthesis and Study of in Vitro Cytotoxic ActivitiesDocument7 pagesNovel Hybrid Molecules of Isoxazole Chalcone Derivatives: Synthesis and Study of in Vitro Cytotoxic ActivitiesRatnakaram Venkata NadhNo ratings yet
- Selectivity Index of Alpinia Galanga Extract and 1'-Acetoxychavicol Acetate On Cancer Cell LinesDocument6 pagesSelectivity Index of Alpinia Galanga Extract and 1'-Acetoxychavicol Acetate On Cancer Cell LinesasrilNo ratings yet
- Ravikanth3 PDFDocument7 pagesRavikanth3 PDFMekala LakshmanNo ratings yet
- Effect of Spinosad and Imidacloprid On Serum Biochemical Alterations in PDFDocument7 pagesEffect of Spinosad and Imidacloprid On Serum Biochemical Alterations in PDFMekala LakshmanNo ratings yet
- Design Synthesis and Evaluation of Genistein Polyamine Conjugates As Multi Functional Anti Alzheimer Agents PDFDocument7 pagesDesign Synthesis and Evaluation of Genistein Polyamine Conjugates As Multi Functional Anti Alzheimer Agents PDFIT InventoryNo ratings yet
- 134 Oxa 1Document5 pages134 Oxa 1P RADHIKA R PRABHUNo ratings yet
- 1 s2.0 S1878DEEEDocument12 pages1 s2.0 S1878DEEEWalid EbaiedNo ratings yet
- 2016 - Inflammopharmacology - Sokmen Et Al - The Antioxidant Activity of Some CurcuminoidsDocument6 pages2016 - Inflammopharmacology - Sokmen Et Al - The Antioxidant Activity of Some CurcuminoidsNadia ErlinaNo ratings yet
- PlantsDocument19 pagesPlantsRahul RanaNo ratings yet
- s12248-024-00899-6Document12 pagess12248-024-00899-6gskcl429No ratings yet
- An in Vitro Study On Antiproliferative and Antiestrogenic Effects ofDocument5 pagesAn in Vitro Study On Antiproliferative and Antiestrogenic Effects ofJose Alberto PbNo ratings yet
- FacileDocument12 pagesFacilesunaina agarwalNo ratings yet
- Articulo FarmoDocument11 pagesArticulo FarmoMiroku SenshiNo ratings yet
- Isolation and Characterization of B-SitosterolDocument6 pagesIsolation and Characterization of B-Sitosterolnirajvyas4meNo ratings yet
- Bioorganic Chemistry: SciencedirectDocument19 pagesBioorganic Chemistry: SciencedirectJUAN DIEGO TRUJILLO ROJASNo ratings yet
- Araújo 2011 S FiliformisDocument9 pagesAraújo 2011 S FiliformisLuizaNo ratings yet
- Anticancer Effects of Cenchrus ciliaris L. on Several Cancer Cell LinesDocument4 pagesAnticancer Effects of Cenchrus ciliaris L. on Several Cancer Cell LinesniaNo ratings yet
- Dr. R. Baskaran - Final KVK Manuscript Draft PDFDocument31 pagesDr. R. Baskaran - Final KVK Manuscript Draft PDFDr. K V KhajapeerNo ratings yet
- Synthesis and Anticancer Effects of Pongamol DDocument8 pagesSynthesis and Anticancer Effects of Pongamol Drahulsaini855No ratings yet
- Anti-Tumor Activity of Annona Squamosa Seeds Extract Containing Annonaceous Acetogenin Compounds PDFDocument6 pagesAnti-Tumor Activity of Annona Squamosa Seeds Extract Containing Annonaceous Acetogenin Compounds PDFElizabeth Noel100% (1)
- CarbazoleDocument6 pagesCarbazoleHasan AliraqiNo ratings yet
- 6 PDFDocument6 pages6 PDFAsif Ullah Khan FaryadiNo ratings yet
- 1 s2.0 S2314853516000093 MainDocument12 pages1 s2.0 S2314853516000093 Mainsarah aliNo ratings yet
- Pharma-Ecology: The Occurrence and Fate of Pharmaceuticals and Personal Care Products in the EnvironmentFrom EverandPharma-Ecology: The Occurrence and Fate of Pharmaceuticals and Personal Care Products in the EnvironmentNo ratings yet
- Crystals 11 01161 v2Document11 pagesCrystals 11 01161 v2Walid Ebid ElgammalNo ratings yet
- European Journal of Medicinal ChemistryDocument14 pagesEuropean Journal of Medicinal ChemistryWalid Ebid ElgammalNo ratings yet
- MoleculesDocument10 pagesMoleculesWalid Ebid ElgammalNo ratings yet
- European Journal of Medicinal Chemistry: Research PaperDocument9 pagesEuropean Journal of Medicinal Chemistry: Research PaperWalid Ebid ElgammalNo ratings yet
- Dimethylformamide Dimethyl Acetal As A Building Block in Heterocyclic SynthesisDocument27 pagesDimethylformamide Dimethyl Acetal As A Building Block in Heterocyclic SynthesisWalid Ebid ElgammalNo ratings yet
- Jhet 4366Document13 pagesJhet 4366Walid Ebid ElgammalNo ratings yet
- An Efficient One-Pot Synthesis of N-(1,3-Diphenyl-1H-Pyrazol-5-yl)amidesDocument7 pagesAn Efficient One-Pot Synthesis of N-(1,3-Diphenyl-1H-Pyrazol-5-yl)amidesWalid Ebid ElgammalNo ratings yet
- An Efficient One-Pot Synthesis of N-(1,3-Diphenyl-1H-Pyrazol-5-yl)amidesDocument7 pagesAn Efficient One-Pot Synthesis of N-(1,3-Diphenyl-1H-Pyrazol-5-yl)amidesWalid Ebid ElgammalNo ratings yet
- Dimethylformamide Dimethyl Acetal As A Building Block in Heterocyclic SynthesisDocument27 pagesDimethylformamide Dimethyl Acetal As A Building Block in Heterocyclic SynthesisWalid Ebid ElgammalNo ratings yet
- Pharmaceuticals 14 00984Document23 pagesPharmaceuticals 14 00984Walid Ebid ElgammalNo ratings yet
- Article 2021Document16 pagesArticle 2021Walid Ebid ElgammalNo ratings yet
- Pharmaceuticals: Ye-Mi Kwon, Sou Hyun Kim, Young-Suk Jung and Jae-Hwan KwakDocument22 pagesPharmaceuticals: Ye-Mi Kwon, Sou Hyun Kim, Young-Suk Jung and Jae-Hwan KwakWalid Ebid ElgammalNo ratings yet
- ضDocument11 pagesضWalid Ebid ElgammalNo ratings yet
- Materials 13 01507 v2Document14 pagesMaterials 13 01507 v2Walid Ebid ElgammalNo ratings yet
- 1 s2.0 S004520ىةةننتتت6821001711 mainDocument20 pages1 s2.0 S004520ىةةننتتت6821001711 mainWalid Ebid ElgammalNo ratings yet
- صDocument6 pagesصWalid Ebid ElgammalNo ratings yet
- Organic & Biomolecular ChemistryDocument4 pagesOrganic & Biomolecular ChemistryWalid Ebid ElgammalNo ratings yet
- Synthesis, Spectral Analysis and Biological Evaluation of N-Alkyl/aralkyl/aryl-4-Chlorobenzenesulfonamide DerivativesDocument9 pagesSynthesis, Spectral Analysis and Biological Evaluation of N-Alkyl/aralkyl/aryl-4-Chlorobenzenesulfonamide DerivativesWalid Ebid ElgammalNo ratings yet
- Mun-Hoe Eddy Chan, Karen A. Crouse, M. Ibrahim M. Tahir, Rozita Rosli, Nasir Umar-Tsafe, Andrew R. CowleyDocument9 pagesMun-Hoe Eddy Chan, Karen A. Crouse, M. Ibrahim M. Tahir, Rozita Rosli, Nasir Umar-Tsafe, Andrew R. CowleyWalid Ebid ElgammalNo ratings yet
- ضDocument11 pagesضWalid Ebid ElgammalNo ratings yet
- A Review: Saccharin Discovery, Synthesis, and Applications: Ibn Al Haitham Journal For Pure and Applied ScienceDocument19 pagesA Review: Saccharin Discovery, Synthesis, and Applications: Ibn Al Haitham Journal For Pure and Applied ScienceWalid Ebid ElgammalNo ratings yet
- Zrocl Sio - Catalyzed Synthesis of Bis (Indoles) Via Conjugate Addition of Indole With Electron-Deficient Alkenes in WaterDocument4 pagesZrocl Sio - Catalyzed Synthesis of Bis (Indoles) Via Conjugate Addition of Indole With Electron-Deficient Alkenes in WaterWalid Ebid ElgammalNo ratings yet
- A Review: Saccharin Discovery, Synthesis, and Applications: Ibn Al Haitham Journal For Pure and Applied ScienceDocument19 pagesA Review: Saccharin Discovery, Synthesis, and Applications: Ibn Al Haitham Journal For Pure and Applied ScienceWalid Ebid ElgammalNo ratings yet
- صDocument6 pagesصWalid Ebid ElgammalNo ratings yet
- Microwave Assisted Synthesis of Some New Spiro - (Indole-Thiazolidine) Derivatives: A Green Chemical PathwayDocument8 pagesMicrowave Assisted Synthesis of Some New Spiro - (Indole-Thiazolidine) Derivatives: A Green Chemical PathwayWalid Ebid ElgammalNo ratings yet
- Design, Synthesis and Antifungal Activity of Novel Indole Derivatives Linked With The 1,2,3-Triazole Moiety Via The Cuaac Click ReactionDocument4 pagesDesign, Synthesis and Antifungal Activity of Novel Indole Derivatives Linked With The 1,2,3-Triazole Moiety Via The Cuaac Click ReactionWalid Ebid ElgammalNo ratings yet
- New Metal Complexes of Sulfonamide: Synthesis, Characterization, In-Vitro Anticancer, Anticholinesterase, Antioxidant, and Antibacterial StudiesDocument13 pagesNew Metal Complexes of Sulfonamide: Synthesis, Characterization, In-Vitro Anticancer, Anticholinesterase, Antioxidant, and Antibacterial StudiesWalid Ebid ElgammalNo ratings yet
- Fischer Indole Synthesis Catalyzed by Novel SO H-Functionalized Ionic Liquids in WaterDocument8 pagesFischer Indole Synthesis Catalyzed by Novel SO H-Functionalized Ionic Liquids in WaterWalid Ebid ElgammalNo ratings yet
- Accepted Manuscript: Bioorganic ChemistryDocument31 pagesAccepted Manuscript: Bioorganic ChemistryWalid Ebid ElgammalNo ratings yet
- ACI Formulations LTD - Dec02 - 2022Document35 pagesACI Formulations LTD - Dec02 - 2022Emdadul HuqNo ratings yet
- Comparative Study of Nylon 11 and PCDT PolyesterDocument15 pagesComparative Study of Nylon 11 and PCDT Polyesterنوشاد علیNo ratings yet
- Oouchi 2019Document7 pagesOouchi 2019Victor Hugo GuzmánNo ratings yet
- JEE MAINS - Test 09 - Solution Notes (Chemistry) - JEE MAINS - Test 09 Solution Notes (Chemistry)Document32 pagesJEE MAINS - Test 09 - Solution Notes (Chemistry) - JEE MAINS - Test 09 Solution Notes (Chemistry)Mohit SuaradkarNo ratings yet
- Fatty Acid Catabolism ExplainedDocument15 pagesFatty Acid Catabolism ExplainedMomena SafdarNo ratings yet
- Xiao 2010Document8 pagesXiao 2010vitor_alberto_7No ratings yet
- Lecture Biochemistry of Connective TissueDocument97 pagesLecture Biochemistry of Connective TissueNeha FathimaNo ratings yet
- Acid deposition impacts on sensitive ecosystemsDocument2 pagesAcid deposition impacts on sensitive ecosystemsZara HamiltonNo ratings yet
- Polyacrylonitrile: Daryan Rahman Danyar MohammedDocument6 pagesPolyacrylonitrile: Daryan Rahman Danyar MohammedKMGV 2No ratings yet
- Lactuca Indica-ChemicalDocument8 pagesLactuca Indica-ChemicalDuy Vũ ThànhNo ratings yet
- Elastomeric Impression MaterialsDocument6 pagesElastomeric Impression MaterialsMarlene CasayuranNo ratings yet
- Tle 9 Q2 - E8 - SLMDocument5 pagesTle 9 Q2 - E8 - SLMCaryll BaylonNo ratings yet
- Organic Compounds Naming GuideDocument8 pagesOrganic Compounds Naming GuideAbhinandan MISHRANo ratings yet
- Unit 2 PolymerDocument61 pagesUnit 2 PolymerAayush ChikhalkarNo ratings yet
- Fullerene: Joshi Dev Dushyantbhai (201901304)Document8 pagesFullerene: Joshi Dev Dushyantbhai (201901304)rates100% (1)
- Chemistry Pre Mid TermDocument3 pagesChemistry Pre Mid TermVatsalyaNo ratings yet
- Exploration On Bioactive Properties of Quinoa Protein Hydrolysate and Peptides: A ReviewDocument15 pagesExploration On Bioactive Properties of Quinoa Protein Hydrolysate and Peptides: A ReviewJhon Esteban Herrera LucumiNo ratings yet
- Answer Two Questions From Section A, One Question From Section B, Two Questions From Section C and One Other Question. Section ADocument6 pagesAnswer Two Questions From Section A, One Question From Section B, Two Questions From Section C and One Other Question. Section AKarren MakamureNo ratings yet
- Nurture Test Series / Joint Package Course (Aiot) : Distance Learning ProgrammeDocument11 pagesNurture Test Series / Joint Package Course (Aiot) : Distance Learning ProgrammePrakhar KataraNo ratings yet
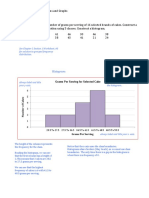
- Histogram:: See Chapter 2 Section 1 Worksheet, #3 For Solution To Grouped Frequency DistributionDocument7 pagesHistogram:: See Chapter 2 Section 1 Worksheet, #3 For Solution To Grouped Frequency DistributionJoanna Paula MagadiaNo ratings yet
- Sigmaline 2000 BaseDocument13 pagesSigmaline 2000 BaseMohamed MahadeerNo ratings yet
- Protein Degradation by The Ubiquitin Proteasome.14Document13 pagesProtein Degradation by The Ubiquitin Proteasome.14007ginniNo ratings yet
- Full Download Test Bank For Organic Chemistry Principles and Mechanisms 2nd Edition Joel Karty PDF Full ChapterDocument36 pagesFull Download Test Bank For Organic Chemistry Principles and Mechanisms 2nd Edition Joel Karty PDF Full Chapterunbackeddealjk2a1100% (18)
- Complete Melon Seed Project Corrected 2Document46 pagesComplete Melon Seed Project Corrected 2Agbede Oluwadamilare BenjaminNo ratings yet
- LectureDocument21 pagesLectureminahilNo ratings yet
- DownloadDocument10 pagesDownloadPepek beloangNo ratings yet
- 10.2478 - Ausal 2019 0002Document21 pages10.2478 - Ausal 2019 0002nekosnow rosecatNo ratings yet
- Enzymes as biological catalysts (E:BCDocument6 pagesEnzymes as biological catalysts (E:BCelena piovesanNo ratings yet