You might also like
- Introduction To BiomarkersDocument83 pagesIntroduction To BiomarkersSubodh NanavatiNo ratings yet
- Drug Discovery Complete NotesDocument5 pagesDrug Discovery Complete NotesSadiqa ForensicNo ratings yet
- Mi RNomicsDocument447 pagesMi RNomicsChalitha somarathneNo ratings yet
- USABO 18 Open Exam - Final Wo AnsDocument17 pagesUSABO 18 Open Exam - Final Wo AnsJ.Chang ChangNo ratings yet
- Advances in de Novo Drug Design - From Conventional To Machine Learning MethodsDocument23 pagesAdvances in de Novo Drug Design - From Conventional To Machine Learning MethodsJean Paul Sarte100% (1)
- Drug Discovery StrategiesDocument5 pagesDrug Discovery Strategiesminad53929 dineroacom100% (1)
- Drug Target Interaction (DTI) and Prediction Using Machine LearningDocument9 pagesDrug Target Interaction (DTI) and Prediction Using Machine LearningIJRASETPublicationsNo ratings yet
- Computer-Aided Drug DesignDocument6 pagesComputer-Aided Drug DesignAnand Barapatre100% (1)
- Bioinformatics in Drug DiscoveryDocument8 pagesBioinformatics in Drug DiscoveryPrincess AleenaNo ratings yet
- 06 - Chapter 1Document37 pages06 - Chapter 1Vikash KushwahaNo ratings yet
- AI in Drug DiscoveryDocument23 pagesAI in Drug Discoverygowtham sai100% (2)
- Role of Computer-Aided Drug Design in Modern Drug DiscoveryDocument16 pagesRole of Computer-Aided Drug Design in Modern Drug DiscoverySourav MajumdarNo ratings yet
- Bel Fiore 2021Document27 pagesBel Fiore 2021Macarena FernandezNo ratings yet
- In Silico Pharmacology For Drug Discovery: Methods For Virtual Ligand Screening and ProfilingDocument12 pagesIn Silico Pharmacology For Drug Discovery: Methods For Virtual Ligand Screening and ProfilingDwi PuspitaNo ratings yet
- Bioinformatics: Original PaperDocument8 pagesBioinformatics: Original PaperLaiq AhmadNo ratings yet
- Chemoinformatics and Chemical Genomics: Potential Utility of in Silico MethodsDocument10 pagesChemoinformatics and Chemical Genomics: Potential Utility of in Silico MethodssuryasanNo ratings yet
- In Silico Pharmacology For Drug Discovery: Applications To Targets and BeyondDocument17 pagesIn Silico Pharmacology For Drug Discovery: Applications To Targets and BeyondAw Yong Yi XiangNo ratings yet
- Drug 10Document10 pagesDrug 10Salem Optimus Technocrates India Private LimitedNo ratings yet
- s12859 017 1638 4 PDFDocument10 pagess12859 017 1638 4 PDFfkamaliyahNo ratings yet
- Gkab 953Document10 pagesGkab 953Kajelcha FikaduNo ratings yet
- Toxicology in Vitro: J.M. Zaldívar Comenges, E. Joossens, J.V. Sala Benito, A. Worth, A. PainiDocument13 pagesToxicology in Vitro: J.M. Zaldívar Comenges, E. Joossens, J.V. Sala Benito, A. Worth, A. PainiDenis EscuderoNo ratings yet
- Waqar Hussain - Bilal Rasheed Machine Learning and Drug DiscoveryDocument4 pagesWaqar Hussain - Bilal Rasheed Machine Learning and Drug DiscoveryWaqar HussainNo ratings yet
- Evidence-Based Absorption, Distribution, Metabolism, Excretion (ADME) and Its Interplay With Alternative Toxicity MethodsDocument16 pagesEvidence-Based Absorption, Distribution, Metabolism, Excretion (ADME) and Its Interplay With Alternative Toxicity MethodsAyi Yurike Tri YantiNo ratings yet
- A Review On Computer Aided Drug Design in Drug Discovery: SJIF Impact Factor 6.647Document13 pagesA Review On Computer Aided Drug Design in Drug Discovery: SJIF Impact Factor 6.647aasthaNo ratings yet
- Semantic Project FFDocument11 pagesSemantic Project FFHabtamu AlebelNo ratings yet
- Drug Target Interaction (DTI) and Prediction Using Machine LearningDocument9 pagesDrug Target Interaction (DTI) and Prediction Using Machine LearningIJRASETPublicationsNo ratings yet
- Aps 2012109Document10 pagesAps 2012109Stym ÅsàtïNo ratings yet
- Drug Discovery Paper m1Document6 pagesDrug Discovery Paper m1Aditi ShimpiNo ratings yet
- FBDD Verview 202008Document7 pagesFBDD Verview 202008fiw ahimNo ratings yet
- Review 1-5 PDFDocument5 pagesReview 1-5 PDFEditor IjprtNo ratings yet
- Conducting Scientific Research Research Hypothesis and Null Hypothesis 2161 0401Document2 pagesConducting Scientific Research Research Hypothesis and Null Hypothesis 2161 0401Zuhra ApriyaniNo ratings yet
- Synergi 3.0Document5 pagesSynergi 3.0lodel1306No ratings yet
- Comprehensive Survey of Recent Drug Discovery UsinDocument37 pagesComprehensive Survey of Recent Drug Discovery UsinCindy GonzálezNo ratings yet
- Deep Learning in Pharmacogenomics: From Gene Regulation To Patient StratificationDocument40 pagesDeep Learning in Pharmacogenomics: From Gene Regulation To Patient StratificationSrikanth ShenoyNo ratings yet
- 1 s2.0 S235291482200034X MainDocument18 pages1 s2.0 S235291482200034X MainpedroNo ratings yet
- Predicting Clinically Promising Therapeutic Hypotheses Using Tensor FactorizationDocument12 pagesPredicting Clinically Promising Therapeutic Hypotheses Using Tensor FactorizationKarl GemayelNo ratings yet
- Neodti: Neural Integration of Neighbor Information From A Heterogeneous Network For Discovering New Drug-Target InteractionsDocument8 pagesNeodti: Neural Integration of Neighbor Information From A Heterogeneous Network For Discovering New Drug-Target InteractionsAJAY ANo ratings yet
- Drug Discovery Using Machine Learning and Data AnalysisDocument9 pagesDrug Discovery Using Machine Learning and Data AnalysisIJRASETPublicationsNo ratings yet
- BasedpairedDocument34 pagesBasedpairedRicardo RomeroNo ratings yet
- Term Paper: Various Techniques in Drug SynthesisDocument14 pagesTerm Paper: Various Techniques in Drug SynthesisUmair MazharNo ratings yet
- Doing It RightDocument2 pagesDoing It Rightjojolim18No ratings yet
- DFinder A Novel End-To-End Graph Embedding-Based MDocument10 pagesDFinder A Novel End-To-End Graph Embedding-Based MNealNo ratings yet
- X ROTEI 3Document6 pagesX ROTEI 3api-3811432No ratings yet
- Advancing Drug Discovery Via Artificial Intelligence: Trends in Pharmacological Sciences July 2019Document14 pagesAdvancing Drug Discovery Via Artificial Intelligence: Trends in Pharmacological Sciences July 2019freeretdocsNo ratings yet
- Adverse Outcome Pathway (AOP) Development I: Strategies and PrinciplesDocument9 pagesAdverse Outcome Pathway (AOP) Development I: Strategies and PrinciplesBharat MahajanNo ratings yet
- Combination Drug Testing DeviceDocument24 pagesCombination Drug Testing DeviceTaha AzadNo ratings yet
- Accepted ManuscriptDocument21 pagesAccepted ManuscriptboniatNo ratings yet
- Link Prediction Drug DiseaseDocument21 pagesLink Prediction Drug Disease20102076No ratings yet
- Regulatory Toxicology and Pharmacology: Survey ReportDocument7 pagesRegulatory Toxicology and Pharmacology: Survey ReportisabellaNo ratings yet
- Application of in Silico Approaches To Predicting Drug-Drug InteractionsDocument5 pagesApplication of in Silico Approaches To Predicting Drug-Drug InteractionsamanuipsNo ratings yet
- Journal of Occupational Medicine and ToxicologyDocument11 pagesJournal of Occupational Medicine and ToxicologyAngélica Rueda RuedaNo ratings yet
- Artificial Intelligence in Drug Discovery: Applications and TechniquesDocument65 pagesArtificial Intelligence in Drug Discovery: Applications and Techniqueszare22No ratings yet
- Biology Project On Ai in MedicineDocument10 pagesBiology Project On Ai in Medicinejapmansingh45678No ratings yet
- Milta 36Document7 pagesMilta 36Victor RoticivNo ratings yet
- Nihms 1681039Document26 pagesNihms 1681039SBTSRIRAMNo ratings yet
- 2023 02 07 23285407v1 Full-2Document19 pages2023 02 07 23285407v1 Full-2Karl GemayelNo ratings yet
- Drug Discovery and DevelopmentDocument5 pagesDrug Discovery and DevelopmentarcherselevatorsNo ratings yet
- Computer Aided Drug Design (Cadd) : Naresh.T M.Pharmacy 1 YR/1 SEMDocument40 pagesComputer Aided Drug Design (Cadd) : Naresh.T M.Pharmacy 1 YR/1 SEMJainendra JainNo ratings yet
- Applying Pharmacogenomics in Drug Development: Call For Collaborative EffortsDocument5 pagesApplying Pharmacogenomics in Drug Development: Call For Collaborative EffortsVeronica BalanNo ratings yet
- Provisional: Molecular Sets (MOSES) : A Benchmarking Platform For Molecular Generation ModelsDocument22 pagesProvisional: Molecular Sets (MOSES) : A Benchmarking Platform For Molecular Generation ModelsRonaldo PaxDeorumNo ratings yet
- Tools 5Document13 pagesTools 5Laiq AhmadNo ratings yet
- This Content Downloaded From 39.41.147.101 On Thu, 27 May 2021 00:05:54 UTCDocument4 pagesThis Content Downloaded From 39.41.147.101 On Thu, 27 May 2021 00:05:54 UTCIrfan Ullah 116-FBAS/MSCHM/F20No ratings yet
- Explained - 2020 Nobel Prize in Chemistry - The HinduDocument4 pagesExplained - 2020 Nobel Prize in Chemistry - The HinduIrfan Ullah 116-FBAS/MSCHM/F20No ratings yet
- Conformationally Directed Drug Design. Peptides and Nucleic Acids As Templates or Targets (PDFDrive)Document268 pagesConformationally Directed Drug Design. Peptides and Nucleic Acids As Templates or Targets (PDFDrive)Irfan Ullah 116-FBAS/MSCHM/F20No ratings yet
- Aguila 2013Document8 pagesAguila 2013Irfan Ullah 116-FBAS/MSCHM/F20No ratings yet
- Written Examination: MOL3005 Immunology: Tuesday June 4 2012, 9.00 Am - 1.00 PMDocument3 pagesWritten Examination: MOL3005 Immunology: Tuesday June 4 2012, 9.00 Am - 1.00 PMsazzad joyNo ratings yet
- Solution Brown Solution: Sample Used Time Oxidized Apple Banana Potato GuavaDocument7 pagesSolution Brown Solution: Sample Used Time Oxidized Apple Banana Potato GuavaLaelannie MagpayoNo ratings yet
- Lesson 2 Genetic Code KESDocument19 pagesLesson 2 Genetic Code KESMaliha RiazNo ratings yet
- General Western Blot ProtocolDocument7 pagesGeneral Western Blot ProtocolMOHAMMAD NAZMUL ISLAMNo ratings yet
- Halder2020 hydrophilicAminoAcidsProteinEthanolDocument10 pagesHalder2020 hydrophilicAminoAcidsProteinEthanolSBNo ratings yet
- Recap:: Questions Based On Previous LectureDocument39 pagesRecap:: Questions Based On Previous LectureMehrin KabirNo ratings yet
- TMP A7 DEDocument26 pagesTMP A7 DEFrontiersNo ratings yet
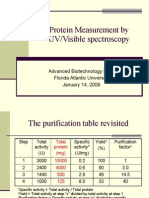
- Protein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Document10 pagesProtein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Gina ZhouNo ratings yet
- SET - 1 Common P.G. Entrance Test 2021 Subject - BotanyDocument9 pagesSET - 1 Common P.G. Entrance Test 2021 Subject - BotanyAlimashree DeepNo ratings yet
- International Journal of VirologyDocument13 pagesInternational Journal of VirologyRaja RonaldoNo ratings yet
- High Throughput Next Generation SequencingDocument2 pagesHigh Throughput Next Generation SequencingJad AwadNo ratings yet
- DNA Replication Is Semi-ConservativeDocument19 pagesDNA Replication Is Semi-ConservativeEuphoriaNo ratings yet
- NLRP3 Inflammasome and Its Inhibitors: A Review: Bo-Zong Shao, Zhe-Qi Xu, Bin-Ze Han, Ding-Feng Su and Chong LiuDocument9 pagesNLRP3 Inflammasome and Its Inhibitors: A Review: Bo-Zong Shao, Zhe-Qi Xu, Bin-Ze Han, Ding-Feng Su and Chong Liuilc67123No ratings yet
- GENE EXPRESSION PROFILE OF PATHOGENICITY FACTORS IN Xylella FastidiosaDocument1 pageGENE EXPRESSION PROFILE OF PATHOGENICITY FACTORS IN Xylella FastidiosacdumenyoNo ratings yet
- Restriction EnzymesDocument14 pagesRestriction EnzymesAman UllahNo ratings yet
- TUBULINADocument8 pagesTUBULINAJuan Florencio Gomez-LeyvaNo ratings yet
- Trieu-Duc Vu, Yuki Iwasaki, Kenshiro Oshima, Ming-Tzu Chiu, Masato Nikaido, Norihiro OkadaDocument12 pagesTrieu-Duc Vu, Yuki Iwasaki, Kenshiro Oshima, Ming-Tzu Chiu, Masato Nikaido, Norihiro OkadaGazza AmrNo ratings yet
- Lab Report 05Document20 pagesLab Report 05DewNo ratings yet
- General Biology 1 - Module 7-MitosisandmeiosisDocument32 pagesGeneral Biology 1 - Module 7-Mitosisandmeiosisエアーア ラシブNo ratings yet
- Chronic Disease Cause by Pseudomonas - AeruginosaDocument22 pagesChronic Disease Cause by Pseudomonas - AeruginosaDhanush andaperumalNo ratings yet
- 5.1.4 Bacterial Morphology Gram Stain InfoDocument7 pages5.1.4 Bacterial Morphology Gram Stain InfoMATTHEW DIAZNo ratings yet
- Ncert Highlight - Cell - by Seep PahujaDocument18 pagesNcert Highlight - Cell - by Seep PahujaskindustrieshelplineNo ratings yet
- Benedicta G. Capunong: Peterpaul - Nacua@deped - Gov.phDocument11 pagesBenedicta G. Capunong: Peterpaul - Nacua@deped - Gov.phMichael MillanesNo ratings yet
- BIOL 315 F2019 Lecture Slides 10-15-2019Document27 pagesBIOL 315 F2019 Lecture Slides 10-15-2019Oliwia NazarukNo ratings yet
- Biol 201 - Exam 2 - KeyDocument7 pagesBiol 201 - Exam 2 - KeyLama ZeinNo ratings yet
- Cell Deaths - Involvement in The Pathogenesis and InterventionDocument21 pagesCell Deaths - Involvement in The Pathogenesis and InterventionSHREYA SENTHIL KUMAR IMS21286No ratings yet
- Understanding Oxidants and Antioxidants: Classical Team With New PlayersDocument13 pagesUnderstanding Oxidants and Antioxidants: Classical Team With New PlayersjenniNo ratings yet
- 3 Molecular Evidence - BioNinjaDocument4 pages3 Molecular Evidence - BioNinjaRosie SunNo ratings yet