You might also like
- 1993 C/K Models Electrical Diagnosis and Wiring DiagramsDocument307 pages1993 C/K Models Electrical Diagnosis and Wiring DiagramsPhil Anderson80% (5)
- Crystal Field Theory - NURDocument5 pagesCrystal Field Theory - NURNurhajrahNo ratings yet
- Chapter 6 Bonding CFTDocument39 pagesChapter 6 Bonding CFTAmirahKamaruddinNo ratings yet
- Crystal Field TheoryDocument6 pagesCrystal Field TheoryRamashish ChoudharyNo ratings yet
- Question Bank in DC CircuitsDocument56 pagesQuestion Bank in DC CircuitsJose Ricardo Garfin Jr.89% (9)
- Module 1 - Electrochemistry (Part 1)Document11 pagesModule 1 - Electrochemistry (Part 1)Steven Lee100% (1)
- Boss GE-10 Service NotesDocument5 pagesBoss GE-10 Service NotesIncubusKissNo ratings yet
- ElectrochemistryDocument58 pagesElectrochemistryWatan SahuNo ratings yet
- Semikron Appl ManualDocument269 pagesSemikron Appl ManualSlim Saloum100% (1)
- Areva&hvdc PDFDocument167 pagesAreva&hvdc PDFAbdallah Salem100% (1)
- Ultrafast Charge Separation From Highly Reductive Znte/Cdse Type Ii Quantum DotsDocument7 pagesUltrafast Charge Separation From Highly Reductive Znte/Cdse Type Ii Quantum DotsIndira DeviNo ratings yet
- 2013-Review-Semiconductor Quantum Dot-Sensitized Solar CellsDocument8 pages2013-Review-Semiconductor Quantum Dot-Sensitized Solar CellsLê Văn TrungNo ratings yet
- Solar Energy Section 13 4 2Document3 pagesSolar Energy Section 13 4 2p KumarNo ratings yet
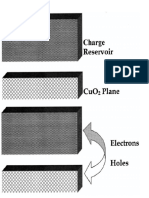
- Electronic Layers in Copper Oxide SuperconductorsDocument1 pageElectronic Layers in Copper Oxide SuperconductorsSJNo ratings yet
- D H A N Ai: or View Them atDocument7 pagesD H A N Ai: or View Them atevsgoud_goudNo ratings yet
- (13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsDocument10 pages(13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsVikash KumarNo ratings yet
- Lithium Air Battery ThesisDocument6 pagesLithium Air Battery Thesisalissacruzomaha100% (2)
- J. Electrochem. Soc. 2016 Cho D428 33Document6 pagesJ. Electrochem. Soc. 2016 Cho D428 33Quý ĐenNo ratings yet
- F 06 S13c EcellmodelsDocument5 pagesF 06 S13c EcellmodelsHatdogNo ratings yet
- 49 IkeriHI 2 PDFDocument6 pages49 IkeriHI 2 PDFMohamed JaawaneNo ratings yet
- Wei Et Al-2010-Journal of Chemical Technology and BiotechnologyDocument6 pagesWei Et Al-2010-Journal of Chemical Technology and BiotechnologyMuhammad Bilal QadirNo ratings yet
- Physics Investigatory Project++Document14 pagesPhysics Investigatory Project++saifahNo ratings yet
- Materials Science and Technology Division "Singlet-Triplet Qubits in Silicon Quantum Dots"Document1 pageMaterials Science and Technology Division "Singlet-Triplet Qubits in Silicon Quantum Dots"cbs78No ratings yet
- Copper Deposition in A Trench: Created in COMSOL Multiphysics 5.5Document16 pagesCopper Deposition in A Trench: Created in COMSOL Multiphysics 5.5EMNo ratings yet
- Quantum Dots For PhotovoltaicDocument4 pagesQuantum Dots For Photovoltaichaddig8No ratings yet
- Electrical Behaviour and Conduction in Cadmium Oxalate Single CrystalsDocument4 pagesElectrical Behaviour and Conduction in Cadmium Oxalate Single CrystalsphysicsjournalNo ratings yet
- Electrochemical Growth and Characterization of Size-Quantized CdTe Thin FilmsDocument6 pagesElectrochemical Growth and Characterization of Size-Quantized CdTe Thin FilmsEdgar Fabian Pinzon NietoNo ratings yet
- Electrical Engg. Dept., Sarvajanik College of Engg. and Tech., Athwalines, Surat, 390 001 Gujarat, India Elect. Engg. Dept., SVNIT, Ichchhanath, Surat, 395 007 Gujarat, IndiaDocument3 pagesElectrical Engg. Dept., Sarvajanik College of Engg. and Tech., Athwalines, Surat, 390 001 Gujarat, India Elect. Engg. Dept., SVNIT, Ichchhanath, Surat, 395 007 Gujarat, Indiacbs78No ratings yet
- IMP-Qdsc Withour FabricationDocument9 pagesIMP-Qdsc Withour FabricationAbhigyan GangulyNo ratings yet
- Quantum Dot Solar CellsDocument19 pagesQuantum Dot Solar Cellsanirbangorain9331No ratings yet
- CQDs OrrDocument9 pagesCQDs OrrPradeep Kumar Yadav Res. Scholar., Dept. of Chemistry, IIT (BHU)No ratings yet
- Hu 2016Document10 pagesHu 2016pramodNo ratings yet
- Band-Selective Holstein Polaron in Luttinger Liquid Material A Moo (A K, RB)Document7 pagesBand-Selective Holstein Polaron in Luttinger Liquid Material A Moo (A K, RB)Scribd ScribdNo ratings yet
- Activity Wenzel Text Voltammetric MethodsDocument15 pagesActivity Wenzel Text Voltammetric MethodsLucica BarbesNo ratings yet
- CDS Modified Cu2O Octahedral Nano-Heterojunction and ItsDocument6 pagesCDS Modified Cu2O Octahedral Nano-Heterojunction and Itsqaisarshah.physicistNo ratings yet
- Experiment 8 ElectrochemistryDocument4 pagesExperiment 8 ElectrochemistryRhett Adrian Seduco0% (1)
- 1 s2.0 S1452398123104172 MainDocument7 pages1 s2.0 S1452398123104172 Main13408169705No ratings yet
- Lecture 23 PDFDocument29 pagesLecture 23 PDFRachit ShahNo ratings yet
- Modelo de Capacitacion Pedagogica para PDocument16 pagesModelo de Capacitacion Pedagogica para PemmanuelNo ratings yet
- Size-Dependent Energy Transfer Between Cdse/Zns Quantum Dots and Gold NanoparticlesDocument5 pagesSize-Dependent Energy Transfer Between Cdse/Zns Quantum Dots and Gold Nanoparticlessouryaya duttaNo ratings yet
- Capacitive Deionization CDI For DesalinationDocument20 pagesCapacitive Deionization CDI For DesalinationFatma Al Belushi100% (1)
- Lecture 8 - The Spectrochemical Series - Color and MagnetismDocument11 pagesLecture 8 - The Spectrochemical Series - Color and MagnetismKimberly RenaeNo ratings yet
- A Mathematical Model of An Electrochemical Capacitor With Double-Layer and Faradaic ProcessesDocument8 pagesA Mathematical Model of An Electrochemical Capacitor With Double-Layer and Faradaic ProcessesArpan KunduNo ratings yet
- Derk 17munich-1 PDFDocument4 pagesDerk 17munich-1 PDFphtvNo ratings yet
- Electrochemical Cell LabDocument9 pagesElectrochemical Cell Labribots0% (1)
- Che 410: Transition Metal Chemistry: Course InstructorDocument24 pagesChe 410: Transition Metal Chemistry: Course InstructorDouglasNo ratings yet
- 10 Chapter Electrochemistry Short Question With Answers PDFDocument11 pages10 Chapter Electrochemistry Short Question With Answers PDFMARITIM GEOFFREY KIPLANGATNo ratings yet
- GROMADSKYI2016 Article CharacterizationOfAgAg2SO4SystDocument7 pagesGROMADSKYI2016 Article CharacterizationOfAgAg2SO4SystlondemonNo ratings yet
- Experiment 8 PhyChem IIDocument5 pagesExperiment 8 PhyChem IIティン ヨロベ100% (1)
- Lundstrom & Stenberg 1974 Charge Injection & Charge Storage in Lipid MultilayersDocument16 pagesLundstrom & Stenberg 1974 Charge Injection & Charge Storage in Lipid MultilayersDr. Uday Sheorey, Ph.D.No ratings yet
- Electrical Study of Thin Film Al/N-Cds Schottky JunctionDocument9 pagesElectrical Study of Thin Film Al/N-Cds Schottky JunctionAnonymous cYpEVvoNo ratings yet
- 2001 BESSY BorisenkoDocument2 pages2001 BESSY BorisenkoXan TolusNo ratings yet
- Effect of Chemical Inhomogeneity in Bismuth-Based Copper Oxide SuperconductorsDocument8 pagesEffect of Chemical Inhomogeneity in Bismuth-Based Copper Oxide SuperconductorsmuhammadNo ratings yet
- Optical Spectra of Divalent Cobalt ComplexesDocument10 pagesOptical Spectra of Divalent Cobalt ComplexesEUGENNo ratings yet
- CuAlO2 Electronic StructureDocument8 pagesCuAlO2 Electronic StructureSanthosh Kumar ChittipoluNo ratings yet
- Electro Chemistry (MS)Document208 pagesElectro Chemistry (MS)Kaustubh SreekharNo ratings yet
- Akimov 2013Document70 pagesAkimov 2013edupibosNo ratings yet
- (2021) Unveiling The Synergistic Inhibition of CL - Cooper Plating: Pivotal Roles of Adsorption and DesorptionDocument8 pages(2021) Unveiling The Synergistic Inhibition of CL - Cooper Plating: Pivotal Roles of Adsorption and Desorptionpeter8324No ratings yet
- Photovoltaic Effect of Copper Photovoltaic Cells: Catibog, J.M.R., Mercado, A.R.T., See, A.J.GDocument3 pagesPhotovoltaic Effect of Copper Photovoltaic Cells: Catibog, J.M.R., Mercado, A.R.T., See, A.J.GRomina MercadoNo ratings yet
- MCLC Taylor and FrancisDocument11 pagesMCLC Taylor and FrancisОлександр КовальчукNo ratings yet
- Coordination CompoundDocument87 pagesCoordination CompoundcskirithikNo ratings yet
- SdmnotesDocument2 pagesSdmnotesSatya SureshNo ratings yet
- Single Electron Transfer Device (Setd)Document17 pagesSingle Electron Transfer Device (Setd)Krisumraj PurkaitNo ratings yet
- Solar Cells Redefined The Promise of Quantum Dot SensitizationFrom EverandSolar Cells Redefined The Promise of Quantum Dot SensitizationNo ratings yet
- Lecture 3 I&M Electromechanical Indicating Instrument 1Document18 pagesLecture 3 I&M Electromechanical Indicating Instrument 1Asad Saeed0% (1)
- OPAMP by COEP ProfDocument171 pagesOPAMP by COEP Profonkarsinare1No ratings yet
- Ieee PapersDocument17 pagesIeee PaperspreddyrkNo ratings yet
- Csa1670087366 Fes0268a-0259a-0263a Ze00Document25 pagesCsa1670087366 Fes0268a-0259a-0263a Ze00RONALTUTILLONo ratings yet
- Presostatos MP55Document6 pagesPresostatos MP55Miguel CallataNo ratings yet
- David EsseiniDocument10 pagesDavid Esseiniশুভ MitraNo ratings yet
- LAB 2 ControlDocument24 pagesLAB 2 ControlfarisNo ratings yet
- Intro To Electronic Comm - Part2Document21 pagesIntro To Electronic Comm - Part2Wawan NazriNo ratings yet
- Saudi Aramco Inspection ChecklistDocument8 pagesSaudi Aramco Inspection ChecklistYehia FelifelNo ratings yet
- Analog Panel & Switchboard Meters - Introduction: General SpecificationsDocument2 pagesAnalog Panel & Switchboard Meters - Introduction: General SpecificationsAnonymous SDeSP1No ratings yet
- Chapter 9Document24 pagesChapter 9Deivasigamani SubramaniyanNo ratings yet
- EF-132 IE Basic Electrical Engineering CourseDocument6 pagesEF-132 IE Basic Electrical Engineering CourseGul sher BalochNo ratings yet
- CSC-121 Breaker Protection IED Engineering and Operation Manual - V1.00Document221 pagesCSC-121 Breaker Protection IED Engineering and Operation Manual - V1.00mentongNo ratings yet
- C-Cores PDFDocument32 pagesC-Cores PDFbpchimeraNo ratings yet
- An Enhanced Droop Control Method For Accurate Load Sharing and Voltage Improvement of Isolated and Interconnected DC MicrogridsDocument11 pagesAn Enhanced Droop Control Method For Accurate Load Sharing and Voltage Improvement of Isolated and Interconnected DC MicrogridsRiad TifaNo ratings yet
- 2019/9/9 Sales-Indo Sales-Indo Sales-Indo PN Ver:02: PreliminaryDocument4 pages2019/9/9 Sales-Indo Sales-Indo Sales-Indo PN Ver:02: PreliminaryCV buanaindahNo ratings yet
- Clutch Coupling Heavy Duty: SFC-500 Bearing MountedDocument2 pagesClutch Coupling Heavy Duty: SFC-500 Bearing MountedElectromateNo ratings yet
- Wharfadale SVP-X Users ManualDocument12 pagesWharfadale SVP-X Users ManualrdbassesNo ratings yet
- Review Letters F: Physic:AiDocument4 pagesReview Letters F: Physic:AiPavel Castro-VillarrealNo ratings yet
- Unit Iv Adc & Dac 2 MarksDocument5 pagesUnit Iv Adc & Dac 2 MarkssasirekhaNo ratings yet
- VVVF - Working Principle & Its OperationDocument18 pagesVVVF - Working Principle & Its Operationdownload4sumitNo ratings yet
- Antenna and Wave Propagation - G. S. N. Raju PDFDocument95 pagesAntenna and Wave Propagation - G. S. N. Raju PDFY Shak KmNo ratings yet
- Finder - 41 Series Relay 0010Document4 pagesFinder - 41 Series Relay 0010Funda HandasNo ratings yet
- DS276 Low Power Transceiver Chip: Features Pin AssignmentDocument11 pagesDS276 Low Power Transceiver Chip: Features Pin AssignmentJairo PadronNo ratings yet