You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Pitch AnythingDocument8 pagesPitch AnythingDoland drumb100% (1)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- 009 Attached-1 NAVFAC P-445 Construction Quality Management PDFDocument194 pages009 Attached-1 NAVFAC P-445 Construction Quality Management PDFSor sopanharithNo ratings yet
- Auditing Multiple Choice Questions and Answers MCQs Auditing MCQ For CA, CS and CMA Exams Principle of Auditing MCQsDocument30 pagesAuditing Multiple Choice Questions and Answers MCQs Auditing MCQ For CA, CS and CMA Exams Principle of Auditing MCQsmirjapur0% (1)
- Revised PARA Element2 Radio LawsDocument81 pagesRevised PARA Element2 Radio LawsAurora Pelagio Vallejos100% (4)
- CBLM - Interpreting Technical DrawingDocument18 pagesCBLM - Interpreting Technical DrawingGlenn F. Salandanan89% (45)
- Leadership Nursing and Patient SafetyDocument172 pagesLeadership Nursing and Patient SafetyRolena Johnette B. PiñeroNo ratings yet
- Gallagher 2014 in Situ Diffraction of Highly DispeDocument11 pagesGallagher 2014 in Situ Diffraction of Highly DispeRaian Yousuf TanmoyNo ratings yet
- Zhou Et Al. Au - CeO2 Series - 2015Document9 pagesZhou Et Al. Au - CeO2 Series - 2015Raian Yousuf TanmoyNo ratings yet
- Surface Density Dependent Catalytic Activity of Single Palladium Atoms Supported On CeriaDocument7 pagesSurface Density Dependent Catalytic Activity of Single Palladium Atoms Supported On CeriaRaian Yousuf TanmoyNo ratings yet
- Urmes-2019-Kinetic-study-of-the-selective-hydrogenation of AcetyleneDocument13 pagesUrmes-2019-Kinetic-study-of-the-selective-hydrogenation of AcetyleneRaian Yousuf Tanmoy100% (1)
- Low-T Hydrocracking of N-HeptaneDocument10 pagesLow-T Hydrocracking of N-HeptaneRaian Yousuf TanmoyNo ratings yet
- Schbib 1996 Kinetics of Front End Acetylene HydrogenationDocument10 pagesSchbib 1996 Kinetics of Front End Acetylene HydrogenationRaian Yousuf TanmoyNo ratings yet
- Wentworth-2016-Experimental-detailed-and-global-kinetics - NO OxidationDocument20 pagesWentworth-2016-Experimental-detailed-and-global-kinetics - NO OxidationRaian Yousuf TanmoyNo ratings yet
- Palladium Acetate Trimer - Understanding Its Ligand-Induced Dissociation Thermochemistry AMKDocument10 pagesPalladium Acetate Trimer - Understanding Its Ligand-Induced Dissociation Thermochemistry AMKRaian Yousuf TanmoyNo ratings yet
- Structural Evolution of Atomically Dispersed PT Catalysts - P Christopher Nat MatDocument7 pagesStructural Evolution of Atomically Dispersed PT Catalysts - P Christopher Nat MatRaian Yousuf TanmoyNo ratings yet
- (Done) Liotta2010 - Silica-Supported Ceria - Effect of Ceria LoadingDocument4 pages(Done) Liotta2010 - Silica-Supported Ceria - Effect of Ceria LoadingRaian Yousuf TanmoyNo ratings yet
- Lu 2021 PT - CeO2Document15 pagesLu 2021 PT - CeO2Raian Yousuf TanmoyNo ratings yet
- Final Detailed Fermenter DesignDocument24 pagesFinal Detailed Fermenter DesignRaian Yousuf TanmoyNo ratings yet
- Hill Second Edition Problem 6.8Document6 pagesHill Second Edition Problem 6.8Raian Yousuf TanmoyNo ratings yet
- Decision Tree AlgorithmDocument22 pagesDecision Tree Algorithmvani_V_prakashNo ratings yet
- A Project Diary-Wps OfficeDocument4 pagesA Project Diary-Wps OfficeSameer ShaikhNo ratings yet
- Blackstone The Dash Model #1610 Owner's ManualDocument53 pagesBlackstone The Dash Model #1610 Owner's ManualSydney Adam SteeleNo ratings yet
- GSM BSC6000 Performance StatisticsDocument72 pagesGSM BSC6000 Performance StatisticsAli AlshwalNo ratings yet
- Unit 1: Power and Responsibility: 1. Important Leadership QualitiesDocument6 pagesUnit 1: Power and Responsibility: 1. Important Leadership QualitiesTrần Thanh MinhNo ratings yet
- BS EN 50483-6-2009 EnglishDocument27 pagesBS EN 50483-6-2009 EnglishДмитро Денис100% (2)
- Directorate of Technical Education, Maharashtra State, MumbaiDocument57 pagesDirectorate of Technical Education, Maharashtra State, MumbaiShubham DahatondeNo ratings yet
- Concise Beam DemoDocument33 pagesConcise Beam DemoluciafmNo ratings yet
- M4110 Leakage Reactance InterfaceDocument2 pagesM4110 Leakage Reactance InterfaceGuru MishraNo ratings yet
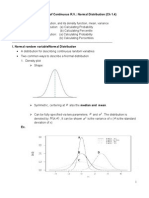
- Distribution of Continuous R.V.: Normal Distribution (CH 1.4) TopicsDocument7 pagesDistribution of Continuous R.V.: Normal Distribution (CH 1.4) TopicsPhạm Ngọc HòaNo ratings yet
- Sample TRM All Series 2020v1 - ShortseDocument40 pagesSample TRM All Series 2020v1 - ShortseSuhail AhmadNo ratings yet
- Dashrath Nandan JAVA (Unit2) NotesDocument18 pagesDashrath Nandan JAVA (Unit2) NotesAbhinandan Singh RanaNo ratings yet
- Bleed Valve FunctionDocument7 pagesBleed Valve FunctionUWT10No ratings yet
- Day 2 - Evident's Official ComplaintDocument18 pagesDay 2 - Evident's Official ComplaintChronicle Herald100% (1)
- IJISRT23JUL645Document11 pagesIJISRT23JUL645International Journal of Innovative Science and Research TechnologyNo ratings yet
- CNNPX310R-6P: General SpecificationsDocument5 pagesCNNPX310R-6P: General SpecificationsZoheir KacimiNo ratings yet
- SPWM Vs SVMDocument11 pagesSPWM Vs SVMpmbalajibtechNo ratings yet
- Re BuyerDocument20 pagesRe BuyerElias OjuokNo ratings yet
- (Database Management Systems) : Biag, Marvin, B. BSIT - 202 September 6 2019Document7 pages(Database Management Systems) : Biag, Marvin, B. BSIT - 202 September 6 2019Marcos JeremyNo ratings yet
- Geology, Logging, Drilling ReportDocument53 pagesGeology, Logging, Drilling Reportwisam alkhooryNo ratings yet
- GeminiDocument397 pagesGeminiJohnnyJC86No ratings yet
- BIO122 - CHAPTER 7 Part 1Document53 pagesBIO122 - CHAPTER 7 Part 1lili100% (1)
- Assessment PN1096617Document14 pagesAssessment PN1096617Amr TarekNo ratings yet
- Civil Engineering Literature Review SampleDocument6 pagesCivil Engineering Literature Review Sampleea442225100% (1)