You might also like
- Hrvoj Vančik (Auth.) - Basic Organic Chemistry For The Life Sciences-Springer International Publishing (2014) PDFDocument188 pagesHrvoj Vančik (Auth.) - Basic Organic Chemistry For The Life Sciences-Springer International Publishing (2014) PDFPako GomezNo ratings yet
- Peptidomimetics FINAL PDFDocument21 pagesPeptidomimetics FINAL PDFVissarapu NagalakshmiNo ratings yet
- Org Chem Expt 7Document4 pagesOrg Chem Expt 7LoveLy GaiLa TabjanNo ratings yet
- OH H R OH H R OH H: FavoredDocument5 pagesOH H R OH H R OH H: FavoredVaibhav TarkasbandNo ratings yet
- Oxoacids of Chlorine by H To O ChemistryDocument44 pagesOxoacids of Chlorine by H To O ChemistryRitu JoharNo ratings yet
- 1289 - Alcohol, Phenol EtherDocument37 pages1289 - Alcohol, Phenol EtherKhan DildarNo ratings yet
- Alcohols Ethers and - 01-TheoryDocument59 pagesAlcohols Ethers and - 01-TheoryRaju SinghNo ratings yet
- FenolDocument43 pagesFenolRedyNo ratings yet
- Experiment 65 Notes: Abbreviations UsedDocument4 pagesExperiment 65 Notes: Abbreviations UsedDIey ChokiEyNo ratings yet
- PhenolsDocument11 pagesPhenolsSai Sasivardhan GampaNo ratings yet
- Alcohols 2Document4 pagesAlcohols 2Abdullah SalmainNo ratings yet
- Carbonyls - Alpha SubstitutionsDocument8 pagesCarbonyls - Alpha SubstitutionsRavi Kiran KoduriNo ratings yet
- Biosynthesis of Natural Products Derived From Shikimic Acid 4.1. Phenyl-Propanoid Natural Products (C - C)Document22 pagesBiosynthesis of Natural Products Derived From Shikimic Acid 4.1. Phenyl-Propanoid Natural Products (C - C)Preeti YadavNo ratings yet
- Alcohol ChemistryDocument73 pagesAlcohol ChemistryBapu Thorat0% (1)
- Topic 5 - AlcoholsDocument7 pagesTopic 5 - AlcoholsRichard WalkerNo ratings yet
- Vdocuments - MX Phenylpropanoids-1Document30 pagesVdocuments - MX Phenylpropanoids-1aninditafaha310No ratings yet
- Chapter17 UclassDocument49 pagesChapter17 Uclass배석우No ratings yet
- Fenil Propanoid Kba 2022 FixDocument44 pagesFenil Propanoid Kba 2022 FixDevi ayuNo ratings yet
- Grupos FuncionalesDocument1 pageGrupos FuncionalesHelena EspañaNo ratings yet
- Phenol: Carboxylation of Phenol: Kolb-Schmitt ReactionDocument9 pagesPhenol: Carboxylation of Phenol: Kolb-Schmitt ReactionX ThrxNo ratings yet
- Hydrophobicity: Physicochemical Properties of Drugs (CH 18)Document35 pagesHydrophobicity: Physicochemical Properties of Drugs (CH 18)afafNo ratings yet
- What Is Organic Chemistry?Document72 pagesWhat Is Organic Chemistry?gs7514424No ratings yet
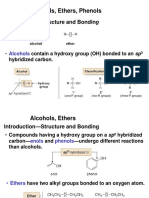
- Alcohols, Ethers, Phenols: - Structure and BondingDocument32 pagesAlcohols, Ethers, Phenols: - Structure and Bondingalshaeri999No ratings yet
- Alcohols and Phenols Class XII NotesDocument74 pagesAlcohols and Phenols Class XII NotesAditya BhattNo ratings yet
- Alcohol Phenol and Ether FinalDocument33 pagesAlcohol Phenol and Ether FinalC.S. KrithikNo ratings yet
- Phenol: Carboxylation of Phenol: Kolb-Schmitt ReactionDocument9 pagesPhenol: Carboxylation of Phenol: Kolb-Schmitt ReactionAkhilaNo ratings yet
- 11-ORGANIC-CHEMISTRY ImpDocument5 pages11-ORGANIC-CHEMISTRY ImpSandeep PlaysNo ratings yet
- Kraft Reactin-Dye SynthesisDocument7 pagesKraft Reactin-Dye Synthesisgulft1777No ratings yet
- PhenolsDocument34 pagesPhenolsLoran Prelya TengayNo ratings yet
- FCH30806 - Phenolics - Lecture 2 - 2024Document47 pagesFCH30806 - Phenolics - Lecture 2 - 2024matthewlowton2255No ratings yet
- Phenols 3 PDFDocument58 pagesPhenols 3 PDFNova sounds - No copyright musicNo ratings yet
- Chapter 7. Alcohols, Phenols, and ThiolsDocument19 pagesChapter 7. Alcohols, Phenols, and Thiolshanna liuNo ratings yet
- Carboxylic Acids, Anhydrides, Esters, and AmidesDocument35 pagesCarboxylic Acids, Anhydrides, Esters, and AmidesHarshwardhan PhatakNo ratings yet
- Experiment 4-Alcohols and PhenolsDocument4 pagesExperiment 4-Alcohols and PhenolsHanieh YousefiNo ratings yet
- 22 - Carboxylic Acids & Esters CORNELLDocument12 pages22 - Carboxylic Acids & Esters CORNELLGeorge SolomouNo ratings yet
- Heterocyclic Compounds: FuranDocument10 pagesHeterocyclic Compounds: FuranSufyan MirzaNo ratings yet
- Phenols: Bio-Organic Chemistry-I BBT-202 Lecture-13Document37 pagesPhenols: Bio-Organic Chemistry-I BBT-202 Lecture-13Sate AhmadNo ratings yet
- Alcohols 2Document22 pagesAlcohols 2Lyana TaylorNo ratings yet
- An Overview of Dietary Polyphenols and Their Therapeutic EffectsDocument15 pagesAn Overview of Dietary Polyphenols and Their Therapeutic EffectsBRUNO JOSE BRITO TEIXEIRANo ratings yet
- Ketones Ethers Esters Alcohols WrksheetsDocument3 pagesKetones Ethers Esters Alcohols WrksheetsPrecious lovely RamosNo ratings yet
- 1288 PhenolsDocument29 pages1288 PhenolsX ThrxNo ratings yet
- CHM 2206-EpoxidesmDocument15 pagesCHM 2206-Epoxidesmkataang445No ratings yet
- An Aldol Condensation To Synthesize ChalconesDocument6 pagesAn Aldol Condensation To Synthesize ChalconesAssyakurNo ratings yet
- 1 CarbohydratesDocument66 pages1 Carbohydratesuswa mansoorNo ratings yet
- Phenol 203Document47 pagesPhenol 203ajibolaakorede20No ratings yet
- Pharmacognosy: Tannin Containing DrugsDocument16 pagesPharmacognosy: Tannin Containing Drugsratchagar aNo ratings yet
- لقطة شاشة 2023-05-27 في 6.17.55 مDocument34 pagesلقطة شاشة 2023-05-27 في 6.17.55 م9c5vtrtqsqNo ratings yet
- Carbohydrates (Monosaccharides) : Anbar University-College of Pharmacy-Clinical Laboratory Sciences Department 2019-2020Document22 pagesCarbohydrates (Monosaccharides) : Anbar University-College of Pharmacy-Clinical Laboratory Sciences Department 2019-2020Zahid Yaqoob BhatNo ratings yet
- Alcohols Phenols PDFDocument88 pagesAlcohols Phenols PDFAnanda VijayasarathyNo ratings yet
- Alcohol OhDocument20 pagesAlcohol OhKara BarbosaNo ratings yet
- Inbound 1668930466231767088Document33 pagesInbound 1668930466231767088urprettygirl14No ratings yet
- XII Zoom PPT Final NewDocument219 pagesXII Zoom PPT Final NewBhuwan JoshiNo ratings yet
- Glucolisis & Fermentación Biología VDocument37 pagesGlucolisis & Fermentación Biología Vcamy.acosta.camposNo ratings yet
- Nomenclature of PhenolsDocument3 pagesNomenclature of PhenolsRockyNo ratings yet
- Chem-Report-OlDocument18 pagesChem-Report-OlThean SivaprahasamNo ratings yet
- ChemicalIdeas13 4OHGroupDocument2 pagesChemicalIdeas13 4OHGroupOCRChemistrySaltersNo ratings yet
- Review - Expt. 3 - Alcohols - PhenolsDocument2 pagesReview - Expt. 3 - Alcohols - PhenolsKaren Joy MagbanuaNo ratings yet
- Removing Organics With IX ResinDocument3 pagesRemoving Organics With IX ResinPrashant RukmangadNo ratings yet
- Recent Developments in the Chemistry of Natural Phenolic Compounds: Proceedings of the Plant Phenolics Group SymposiumFrom EverandRecent Developments in the Chemistry of Natural Phenolic Compounds: Proceedings of the Plant Phenolics Group SymposiumW. D. OllisNo ratings yet
- Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryFrom EverandOrganometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryF. G. A. StoneNo ratings yet
- Organic Chemistry - WikipediaDocument11 pagesOrganic Chemistry - Wikipediatsvmpm1765No ratings yet
- Jawaharlal Nehru University NEW DELHI-110067: Admission - No/3 (Ii) /2019Document7 pagesJawaharlal Nehru University NEW DELHI-110067: Admission - No/3 (Ii) /2019Vikas Kodan KablanaNo ratings yet
- Praktikum Kimia Organik 1 Uin Jakarta PkimDocument31 pagesPraktikum Kimia Organik 1 Uin Jakarta PkimNur BillahNo ratings yet
- Deocument - 282full Download Book Heterocyclic Organic Corrosion Inhibitors Principles and Applications PDFDocument41 pagesDeocument - 282full Download Book Heterocyclic Organic Corrosion Inhibitors Principles and Applications PDFrichard.lamar761100% (31)
- MDSO801D-Understanding Oil and Gas Business-V1FinalDocument350 pagesMDSO801D-Understanding Oil and Gas Business-V1Finalbaggi0% (1)
- Hetro Cyclic Compound NomenclatureDocument8 pagesHetro Cyclic Compound Nomenclatureshq_jsrNo ratings yet
- 9 FCFD 50 B 0 Fa 0 A 3 C 14 CDocument453 pages9 FCFD 50 B 0 Fa 0 A 3 C 14 Cnasrpooya100% (1)
- Rainer Herges - Topology in Chemistry: Designing Mobius MoleculesDocument23 pagesRainer Herges - Topology in Chemistry: Designing Mobius MoleculesOmsadsiNo ratings yet
- Classification IupacDocument32 pagesClassification IupacaneeshvacNo ratings yet
- Wu 2007Document30 pagesWu 2007gjfhkjNo ratings yet
- Heterocycles JapanDocument9 pagesHeterocycles JapanNgô Tuấn KiệtNo ratings yet
- WoodwardDocument21 pagesWoodwardanastasia0% (1)
- GaloreTx-Peptide Chemistry Capabilities Feb 24 2019Document13 pagesGaloreTx-Peptide Chemistry Capabilities Feb 24 2019chidambaramrNo ratings yet
- Morrison and BoydDocument18 pagesMorrison and BoydAman Shrivastava0% (1)
- Benzene Aromatic CompoundsDocument14 pagesBenzene Aromatic CompoundsBook of Life fgfhfghfghfghNo ratings yet
- General Organic Chemistry-01 - TheoryDocument54 pagesGeneral Organic Chemistry-01 - TheoryRaju SinghNo ratings yet
- 10 1021@acs Chemrev 6b00076Document238 pages10 1021@acs Chemrev 6b00076Petru Apostol100% (1)
- Science 2023 Question Paper Set 31-1-1Document16 pagesScience 2023 Question Paper Set 31-1-1aashishj1976No ratings yet
- Us9453128 PDFDocument57 pagesUs9453128 PDFVansala GanesanNo ratings yet
- Acree 1999Document139 pagesAcree 1999valdes2No ratings yet
- Saponins, Classi Cation and Occurrence in The Plant KingdomDocument23 pagesSaponins, Classi Cation and Occurrence in The Plant KingdomyvcgNo ratings yet
- Chem Volume 3Document170 pagesChem Volume 3Marc Laurenze CelisNo ratings yet
- UntitledDocument65 pagesUntitledMoaz AzabNo ratings yet
- Mass Spectroscopy - PPTX Paper 2Document27 pagesMass Spectroscopy - PPTX Paper 2Prerana RaneNo ratings yet
- The Bullvalene Story. The Conception of Bullvalene, A Molecule That Has No Permanent StructureDocument4 pagesThe Bullvalene Story. The Conception of Bullvalene, A Molecule That Has No Permanent StructureThirumalai DNo ratings yet
- The Aromatic RingDocument4 pagesThe Aromatic RingGuadalupeNo ratings yet
- The To Prepare For Only App You NeedDocument146 pagesThe To Prepare For Only App You Needsai mukeshNo ratings yet
- Che 232 Test 1 Sptember 2007Document16 pagesChe 232 Test 1 Sptember 2007BONOLO RANKONo ratings yet