You might also like
- How Great Thou ArtDocument8 pagesHow Great Thou ArtGilbey AguirreNo ratings yet
- Scicent AB TE U9 e 1Document28 pagesScicent AB TE U9 e 1chapmen chan100% (1)
- Design and Construction of Security Door AlarmDocument30 pagesDesign and Construction of Security Door Alarmsanusi bello bakuraNo ratings yet
- Soil AnalysisDocument226 pagesSoil AnalysisAnupriya OberoiNo ratings yet
- Reviewer For Chemical Engineering Licensure Examination 3 Edition Solutions ManualDocument56 pagesReviewer For Chemical Engineering Licensure Examination 3 Edition Solutions ManualSherry Anne Ynciong Panganiban100% (4)
- Synthesis and Characterization of Bismuth Oxide Doped Titanium Dioxide and Its Antibacterial ActivityDocument16 pagesSynthesis and Characterization of Bismuth Oxide Doped Titanium Dioxide and Its Antibacterial ActivityShinta Novita Sari100% (1)
- Analysis of Test Sticks From Surface Testing in The Slovak Republic SFAGIDocument15 pagesAnalysis of Test Sticks From Surface Testing in The Slovak Republic SFAGIsfagiNo ratings yet
- Question Paper June 2023 (H43201)Document28 pagesQuestion Paper June 2023 (H43201)Dominic Afoakwah (Jack)No ratings yet
- General Instrumentation 21.22Document23 pagesGeneral Instrumentation 21.22sanusi bello bakuraNo ratings yet
- M1338-048 Ing AnDocument129 pagesM1338-048 Ing AnFILL TECHNIC MACHINERYNo ratings yet
- ASTM - D2035 19 - en USDocument4 pagesASTM - D2035 19 - en USEtel Del ValleNo ratings yet
- Design and Implementation of A Web Based System For Distance LearningDocument17 pagesDesign and Implementation of A Web Based System For Distance Learningsanusi bello bakura100% (5)
- Herbaceousplants: Cultivation Methods Grazing AND Environmental ImpactsDocument32 pagesHerbaceousplants: Cultivation Methods Grazing AND Environmental ImpactsvikasNo ratings yet
- Droste Chapter 5Document33 pagesDroste Chapter 5Kirito KazutoNo ratings yet
- HUMR 310C JS Zebrafish Assignment, Sara Fares and Habib Rammal-2023Document6 pagesHUMR 310C JS Zebrafish Assignment, Sara Fares and Habib Rammal-202342030570No ratings yet
- OMW 400 BrochureDocument1 pageOMW 400 BrochuresridhardotsharmaNo ratings yet
- React Banner DemoDocument1 pageReact Banner DemoaimeternitystaffNo ratings yet
- Perfect Diet Recipe Ebook PDFDocument24 pagesPerfect Diet Recipe Ebook PDFJohn SowellNo ratings yet
- NationalNCDGuidelineJune102016forprint PDFDocument221 pagesNationalNCDGuidelineJune102016forprint PDFBirhanu JireNo ratings yet
- Fmcowerri, Fami Lymedi CI Ne, Part 1mockexami Nati On, September 2017. (N)Document16 pagesFmcowerri, Fami Lymedi CI Ne, Part 1mockexami Nati On, September 2017. (N)okpala ikennaNo ratings yet
- PosterDocument1 pagePosterwclyne1576No ratings yet
- M01 Mathematics (B1+B2) Rev.00 Pages 11Document10 pagesM01 Mathematics (B1+B2) Rev.00 Pages 11Muhammed MudassirNo ratings yet
- ApurvaBandyopadhyay CV CompressedDocument1 pageApurvaBandyopadhyay CV CompressedApurva BandyopadhyayNo ratings yet
- HOVA Mechanical Seals and Parts V17sDocument36 pagesHOVA Mechanical Seals and Parts V17sLendl dela CruzNo ratings yet
- Bio CH 6Document17 pagesBio CH 6莓乃兹No ratings yet
- Hanging Drop PreparationDocument2 pagesHanging Drop PreparationAlwin JrNo ratings yet
- Complete Notes, Computer Applications, B Pharmacy 2nd Sem, Carewell PharmaDocument38 pagesComplete Notes, Computer Applications, B Pharmacy 2nd Sem, Carewell PharmaRajeev ParmarNo ratings yet
- Important About JuiceDocument9 pagesImportant About JuicemulerstarNo ratings yet
- UntitledDocument1 pageUntitledNikhil SinghNo ratings yet
- Progress Chart and Achievement Chart HarrisDocument15 pagesProgress Chart and Achievement Chart HarrisHarris MalakiNo ratings yet
- 126 - Soal Try Out CBTDocument54 pages126 - Soal Try Out CBTgunawanNo ratings yet
- Orgl 3321 Dddmi ArtifactDocument5 pagesOrgl 3321 Dddmi Artifactapi-673975467No ratings yet
- 15 BCM FNLDocument4 pages15 BCM FNLZaid ZidiNo ratings yet
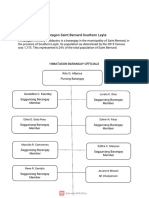
- Hi Mat Agonsai Ntber Nar Dsout Her Nleyt E: Pobl Aci OnDocument7 pagesHi Mat Agonsai Ntber Nar Dsout Her Nleyt E: Pobl Aci Onmaribeth mendozaNo ratings yet
- Aarti GreenBeansDocument1 pageAarti GreenBeansDafne Mysœlf RamirezNo ratings yet
- SUN Lens: Use Descr I PT I On Fi L T Er Cat Egor y TR Ansmi T T AnceDocument1 pageSUN Lens: Use Descr I PT I On Fi L T Er Cat Egor y TR Ansmi T T AnceBiksam CKNo ratings yet
- TurftrucksterDocument263 pagesTurftrucksterGisell ZapataNo ratings yet
- Makalah: MI Crosoftexcel, Fungsifi Tur, Logi KadanmenuDocument13 pagesMakalah: MI Crosoftexcel, Fungsifi Tur, Logi KadanmenuarifNo ratings yet
- Drug Name GENERIC NAME: Ceftriaxone BRAND NAME: RocephinDocument33 pagesDrug Name GENERIC NAME: Ceftriaxone BRAND NAME: RocephinLauren JalandoniNo ratings yet
- Sinonim ObatDocument18 pagesSinonim ObatWin EfendiNo ratings yet
- Sed o N The Findin Gs To Awn F Rom T His ST Udy, Speci Fic Ou Tput C Ould Searc Hers P Roduc E?Document1 pageSed o N The Findin Gs To Awn F Rom T His ST Udy, Speci Fic Ou Tput C Ould Searc Hers P Roduc E?Jeanrose Masisado RaymundoNo ratings yet
- Antimicrobial ResistanceDocument19 pagesAntimicrobial Resistanceapi-19969058No ratings yet
- Consumer Communications: Defining Terms Used in Environmental SustainabilityDocument32 pagesConsumer Communications: Defining Terms Used in Environmental SustainabilityAlma AcostaNo ratings yet
- TF 781587412Document4 pagesTF 781587412ianachieviciNo ratings yet
- All About Me RachelDocument6 pagesAll About Me RacheltconstantineNo ratings yet
- BMH SR PT - MEIDocument1 pageBMH SR PT - MEIarief bayuNo ratings yet
- FI I Tjee: Physi CS, Chemi Stry&Mathemati CSDocument15 pagesFI I Tjee: Physi CS, Chemi Stry&Mathemati CSARARNo ratings yet
- Kolhe Srujan Digital PortfolioDocument8 pagesKolhe Srujan Digital PortfolioShubha JayantNo ratings yet
- S66RE InstructionsDocument2 pagesS66RE InstructionsNazar ChornyiNo ratings yet
- Anamika Kushwaha 12th Result 2023.1st Div?Document1 pageAnamika Kushwaha 12th Result 2023.1st Div?Nitish KushwahaNo ratings yet
- Curriculum VitaeDocument1 pageCurriculum Vitaeandrea prietoNo ratings yet
- Chapter 1 Bio QuestionsDocument4 pagesChapter 1 Bio QuestionsAneesa GulNo ratings yet
- (1240-MC-245) 25713-220-V1A-ECM1-00017 - PDFDocument1 page(1240-MC-245) 25713-220-V1A-ECM1-00017 - PDFAngela CamaggiNo ratings yet
- Q4Tday MAG2009 enDocument32 pagesQ4Tday MAG2009 enJose ManuelNo ratings yet
- Plan AktivnostiDocument5 pagesPlan AktivnostiOptima EnergijaNo ratings yet
- (1240-SG-242) 25713-220-V1A-ESL0-00085 - PDFDocument1 page(1240-SG-242) 25713-220-V1A-ESL0-00085 - PDFAngela CamaggiNo ratings yet
- Data Gathering Instrument For Trainees Characteristics - SubmissionDocument4 pagesData Gathering Instrument For Trainees Characteristics - SubmissionJude MetanteNo ratings yet
- Equipo 6. Actividad ReplicacionDocument3 pagesEquipo 6. Actividad ReplicacionMariajose Barajas MartinezNo ratings yet
- DanDocument1 pageDansean6474No ratings yet
- Board ResultDocument1 pageBoard ResultclevergurujiNo ratings yet
- Brief ProfileDocument1 pageBrief ProfileCzarina Paola IlaganNo ratings yet
- Poster Pepki4 2007Document6 pagesPoster Pepki4 2007feryvera01No ratings yet
- HTP Stab WoundsDocument9 pagesHTP Stab WoundsEYANAH DELOS REYESNo ratings yet
- Mil Whylearnanotherlanguage 073015Document1 pageMil Whylearnanotherlanguage 073015api-248115700No ratings yet
- Clasamentul Mediilor GeneraleDocument2 pagesClasamentul Mediilor GeneraleGabriel CosminNo ratings yet
- Guidance FOR Radio Installations CH A P T e R 2 SurveysDocument10 pagesGuidance FOR Radio Installations CH A P T e R 2 SurveysNguyễn Đức TựNo ratings yet
- Int Roduc T I On: 1) Li Mi T E Dcul T I Vabl Eare ADocument6 pagesInt Roduc T I On: 1) Li Mi T E Dcul T I Vabl Eare ANOOR CHNo ratings yet
- H5E H5M H5X: Illustrated Parts CatalogDocument33 pagesH5E H5M H5X: Illustrated Parts CatalogFrank RosalesNo ratings yet
- Module 11 - Safety and Quality AssuranceDocument38 pagesModule 11 - Safety and Quality Assurancehaider.highlandNo ratings yet
- Java HandoutDocument34 pagesJava Handoutsanusi bello bakuraNo ratings yet
- Advantages and Disadvantages of Sel1Document4 pagesAdvantages and Disadvantages of Sel1sanusi bello bakuraNo ratings yet
- COM 123 Application Packages I TheoryDocument26 pagesCOM 123 Application Packages I Theorysanusi bello bakura100% (2)
- Raw Industrial AssDocument43 pagesRaw Industrial Asssanusi bello bakuraNo ratings yet
- Conditions For Healthy IRDocument5 pagesConditions For Healthy IRsanusi bello bakuraNo ratings yet
- Management Information TechnologyDocument4 pagesManagement Information Technologysanusi bello bakuraNo ratings yet
- Design and Implementation of NYSC Posting SysytemDocument38 pagesDesign and Implementation of NYSC Posting Sysytemsanusi bello bakuraNo ratings yet
- Design and Implementation of Online Clearance SystemDocument8 pagesDesign and Implementation of Online Clearance Systemsanusi bello bakuraNo ratings yet
- Application of HPI (Heavy Metal Pollution Index) and Correlation Coefficient For The Assessment of Ground Water Quality Near Ash Ponds of Thermal Power PlantsDocument11 pagesApplication of HPI (Heavy Metal Pollution Index) and Correlation Coefficient For The Assessment of Ground Water Quality Near Ash Ponds of Thermal Power Plantssanusi bello bakuraNo ratings yet
- Design and Implementation of Allocation of Supervisors To Siwes StudentDocument7 pagesDesign and Implementation of Allocation of Supervisors To Siwes Studentsanusi bello bakura0% (1)
- Finalexercise Methanolprocess 150227020443 Conversion Gate01 PDFDocument6 pagesFinalexercise Methanolprocess 150227020443 Conversion Gate01 PDFMed RjebNo ratings yet
- 5.notes .Dilutions - Colligative.FPD .BPEDocument2 pages5.notes .Dilutions - Colligative.FPD .BPEAmaris HopkinsNo ratings yet
- Assessment of Drought Tolerance in Mung Bean Cultivarslines As Depicted by The Activities of Germination Enzymes, Seedling's Antioxidative Potential and Nutrient AcquisitionDocument33 pagesAssessment of Drought Tolerance in Mung Bean Cultivarslines As Depicted by The Activities of Germination Enzymes, Seedling's Antioxidative Potential and Nutrient AcquisitionFaisal ShehzadNo ratings yet
- Agidol-1: SECTION 1: Identification of The Substance/mixture and of The Company/undertaking 1.1. Product IdentifierDocument24 pagesAgidol-1: SECTION 1: Identification of The Substance/mixture and of The Company/undertaking 1.1. Product IdentifierSekawan CosmeticsNo ratings yet
- Series DS-1 Dry-Type Sprinklers 5.6K Pendent, Upright, and Horizontal Sidewall Quick Response, Standard Coverage General DescriptionDocument10 pagesSeries DS-1 Dry-Type Sprinklers 5.6K Pendent, Upright, and Horizontal Sidewall Quick Response, Standard Coverage General DescriptionRoobertho AthhondoNo ratings yet
- Carboguard 893 SG PDFDocument2 pagesCarboguard 893 SG PDFQA QCNo ratings yet
- Model Paper-7Document13 pagesModel Paper-7Anonymous SOQFPWBNo ratings yet
- Advanced Engineering Materials: Material Safety Data SheetDocument3 pagesAdvanced Engineering Materials: Material Safety Data SheetJason AlexNo ratings yet
- EVE (Energy Very Endure) Primary Batteries 2018Document4 pagesEVE (Energy Very Endure) Primary Batteries 2018MedSparkNo ratings yet
- 07 UhaDocument15 pages07 UhaMatías GarcíaNo ratings yet
- COLLIGDocument3 pagesCOLLIGYokaris JTNo ratings yet
- Neo Fiberglass Mesh 160 GSM TDSDocument1 pageNeo Fiberglass Mesh 160 GSM TDSAkosh AchuNo ratings yet
- Organometallic ChemistryDocument25 pagesOrganometallic ChemistryKaran RavalNo ratings yet
- Introductory Analytical Chemistry CurriculumDocument4 pagesIntroductory Analytical Chemistry CurriculumAcidri AbdulkarimNo ratings yet
- Urine Screening For Metabolic DisordersDocument12 pagesUrine Screening For Metabolic DisordersMitch Ibay100% (1)
- Titalon 6800GF-HT: Charpy Impact Strength (Notched)Document1 pageTitalon 6800GF-HT: Charpy Impact Strength (Notched)katolokchokNo ratings yet
- Metabolism in Fetus and NewbornDocument20 pagesMetabolism in Fetus and NewbornBikash SahNo ratings yet
- SP0387-2014 Formerly RP0387 Metallurgical and Inspection Requirements For Cast Galvanic Anodes For Offshore Applications (21036-SG)Document12 pagesSP0387-2014 Formerly RP0387 Metallurgical and Inspection Requirements For Cast Galvanic Anodes For Offshore Applications (21036-SG)Setiawan WijayantoNo ratings yet
- Technical Bulletin: Wet Processing of 100% Cotton Knitted FabricsDocument11 pagesTechnical Bulletin: Wet Processing of 100% Cotton Knitted FabricsRafayMalikNo ratings yet
- Chemistry 213: Experiment 7 Determining Water Hardness by EDTA TitrationDocument6 pagesChemistry 213: Experiment 7 Determining Water Hardness by EDTA TitrationAvinashNo ratings yet
- OringmatDocument39 pagesOringmatRoby MastreNo ratings yet
- Preparation of Inulin From Chicory Roots: H LianthuDocument5 pagesPreparation of Inulin From Chicory Roots: H LianthuDaniel Patel100% (1)
- FYBSC ChemistryDocument13 pagesFYBSC Chemistryhitech cityNo ratings yet