You might also like
- MOF CatalysisDocument68 pagesMOF CatalysisMarcio VclNo ratings yet
- Acscatal 6b00848Document16 pagesAcscatal 6b00848Vipul SinghNo ratings yet
- Review Solar Driver Matal Halide Perovskite For PhotocatalysisDocument17 pagesReview Solar Driver Matal Halide Perovskite For Photocatalysisआदित्य नारायण सिंहNo ratings yet
- MOF Untuk KatalisDocument11 pagesMOF Untuk KatalisgrafijokerNo ratings yet
- Roles of Cocatalysts in Photocatalysis and PhotoelectrocatalysisDocument10 pagesRoles of Cocatalysts in Photocatalysis and PhotoelectrocatalysisDibyajyoti GhoshNo ratings yet
- Acs JPCC 0c10476Document7 pagesAcs JPCC 0c10476schreier labNo ratings yet
- Engl, Reiser - 2020Document8 pagesEngl, Reiser - 2020franciscaNo ratings yet
- Zhang Et Al 2024 Biomimetic Frustrated Lewis Pair Catalysts For Hydrogenation of Co To Methanol at Low TemperaturesDocument10 pagesZhang Et Al 2024 Biomimetic Frustrated Lewis Pair Catalysts For Hydrogenation of Co To Methanol at Low TemperaturesTEZPURIAN SHANKAR 06091999No ratings yet
- Organic SynthesisDocument9 pagesOrganic SynthesisUmair Tariq RamayNo ratings yet
- Direct H2 SDecompositionby Plasmonic PhotocatalysisDocument9 pagesDirect H2 SDecompositionby Plasmonic PhotocatalysisSyeda Ammara AnwarNo ratings yet
- ACS Catalysis 2020 Engineering Lower Coordination Atoms Onto NiO-Co3ODocument9 pagesACS Catalysis 2020 Engineering Lower Coordination Atoms Onto NiO-Co3OTanveer KhanNo ratings yet
- Molecular Iridium Complexes in Metal Organic Frameworks Catalyze CO Hydrogenation Via Concerted Proton and Hydride TransferDocument4 pagesMolecular Iridium Complexes in Metal Organic Frameworks Catalyze CO Hydrogenation Via Concerted Proton and Hydride TransfergrignardreagnetsNo ratings yet
- Raja 2012Document13 pagesRaja 2012Diana ChiscaNo ratings yet
- Org. Lett. 2022, 24, 25, 4630–4634Document5 pagesOrg. Lett. 2022, 24, 25, 4630–4634NoimurNo ratings yet
- ACS Catalysis - 2015 - Descriptor Analysis in Methanol Conversion On Doped CeO2 (111) Guidelines For Selectivity TuningDocument8 pagesACS Catalysis - 2015 - Descriptor Analysis in Methanol Conversion On Doped CeO2 (111) Guidelines For Selectivity TuningAli HafeezNo ratings yet
- File 20211209 211539 C4cy00296bDocument10 pagesFile 20211209 211539 C4cy00296bNgọc Huyền NguyễnNo ratings yet
- ACS Catalysis - 2015 - Mechanistic Details and Reactivity Descriptors in Oxidation and Acid Catalysis of MethanolDocument17 pagesACS Catalysis - 2015 - Mechanistic Details and Reactivity Descriptors in Oxidation and Acid Catalysis of MethanolAli HafeezNo ratings yet
- John 2010Document24 pagesJohn 2010migenyasuyoshiNo ratings yet
- Review Electrocoagulation - Flotation - 2009Document6 pagesReview Electrocoagulation - Flotation - 2009BibNo ratings yet
- Materials Chemistry A: Journal ofDocument8 pagesMaterials Chemistry A: Journal ofN AgNo ratings yet
- Chem Soc Rev: Review ArticleDocument40 pagesChem Soc Rev: Review ArticleSeptian Perwira YudhaNo ratings yet
- 2014 (Aerobic Homocoupling of Arylboronic Acids Catalysed by Copper Terephthalate Metal-OrganicDocument11 pages2014 (Aerobic Homocoupling of Arylboronic Acids Catalysed by Copper Terephthalate Metal-OrganicHawta AbdullaNo ratings yet
- Review 2Document14 pagesReview 2Shaswata KunduNo ratings yet
- Tetrahedron LettersDocument5 pagesTetrahedron LettersAntônio Neto MachadoNo ratings yet
- ACS Catalysis - 2012 - Pd-Modified Tungsten Carbide For Methanol Electro-Oxidation From Surface Science Studies To Electrochemical EvaluDocument8 pagesACS Catalysis - 2012 - Pd-Modified Tungsten Carbide For Methanol Electro-Oxidation From Surface Science Studies To Electrochemical EvaluAli HafeezNo ratings yet
- Jacs 3c03786Document10 pagesJacs 3c03786Nongnuch ArtrithNo ratings yet
- Acs Jpca 0c09116Document11 pagesAcs Jpca 0c09116Durga Prasad KalamNo ratings yet
- Gusev S Kaya 2003Document7 pagesGusev S Kaya 2003Lim LeepingNo ratings yet
- 4CzIPN T Bu-Catalyzed Proton-Coupled Electron Transfer For Photosynthesis of Phosphorylated N HeteroaromaticsDocument9 pages4CzIPN T Bu-Catalyzed Proton-Coupled Electron Transfer For Photosynthesis of Phosphorylated N HeteroaromaticsLalchan MiahNo ratings yet
- 1 s2.0 S2452223620301115 MainDocument10 pages1 s2.0 S2452223620301115 MainDrawing and Artistic DecorationsNo ratings yet
- Dihydroisoquinoline Acs - Orglett.1c03054Document5 pagesDihydroisoquinoline Acs - Orglett.1c03054Mutiva YyNo ratings yet
- 2013-Chem Soc rev-CO2RRDocument14 pages2013-Chem Soc rev-CO2RRchristopher chenNo ratings yet
- CAPE Biology Syllabus With Specimen Papers - Split - 1Document5 pagesCAPE Biology Syllabus With Specimen Papers - Split - 1Enz JosephNo ratings yet
- CAPE Biology Unit 2 SyllabusDocument20 pagesCAPE Biology Unit 2 Syllabussmithsashay74No ratings yet
- Supported Bifunctional Molybdenum Oxide-Palladium Catalysts For Selective Hydrodeoxygenation of Biomass-Derived Polyols and 1,4-AnhydroerythritolDocument9 pagesSupported Bifunctional Molybdenum Oxide-Palladium Catalysts For Selective Hydrodeoxygenation of Biomass-Derived Polyols and 1,4-AnhydroerythritolAishwarya K NNo ratings yet
- Catala: Perovskite Oxides: MaterDocument8 pagesCatala: Perovskite Oxides: MaterAnonymous G9znMFFRpNo ratings yet
- d2cp05133h PDFDocument16 pagesd2cp05133h PDF魏一琛No ratings yet
- 10 1021@acs Chemrev 9b00638Document55 pages10 1021@acs Chemrev 9b00638Joseph KfouryNo ratings yet
- 立體選擇性探討 丙交酯立體選擇性開環聚合的單中心催化劑Document8 pages立體選擇性探討 丙交酯立體選擇性開環聚合的單中心催化劑hungNo ratings yet
- Ijms 23 12652 v2Document12 pagesIjms 23 12652 v2Deym GómezNo ratings yet
- Ecent Advances of Catalysis in The Hydrogenation andDocument14 pagesEcent Advances of Catalysis in The Hydrogenation andalexis munyentwaliNo ratings yet
- Anie 200300629Document16 pagesAnie 200300629wiam wiamNo ratings yet
- 10 1021@acs Organomet 0c00161Document9 pages10 1021@acs Organomet 0c00161Lakshay KathuriaNo ratings yet
- Low-Temperature Hydrogenation of The C O Bond of Propanal Over Ni-Pt Bimetallic Catalysts: From Model Surfaces To Supported CatalystsDocument7 pagesLow-Temperature Hydrogenation of The C O Bond of Propanal Over Ni-Pt Bimetallic Catalysts: From Model Surfaces To Supported CatalystsAmir DanialNo ratings yet
- Kinetics of Soybean Oil Epoxidation in A Semibatch Reactor: AccessDocument12 pagesKinetics of Soybean Oil Epoxidation in A Semibatch Reactor: AccessCinthia Sierra LgarsNo ratings yet
- @A Review of Catalysts For The Electroreduction of Carbon Dioxide To Produce Low-Carbon FuelsDocument45 pages@A Review of Catalysts For The Electroreduction of Carbon Dioxide To Produce Low-Carbon Fuelschristopher chenNo ratings yet
- LDH Catalysts PDFDocument20 pagesLDH Catalysts PDFAhsan Iqbal AlamgirNo ratings yet
- Yu Et Al 2022 Hydrogenation of Co2 To Methane Over A Ru Rutio2 Surface A DFT Investigation Into The Significant Role ofDocument13 pagesYu Et Al 2022 Hydrogenation of Co2 To Methane Over A Ru Rutio2 Surface A DFT Investigation Into The Significant Role ofpattanapon.kNo ratings yet
- Acs Inorgchem 2c01484Document8 pagesAcs Inorgchem 2c01484Tahir SajjadNo ratings yet
- Applied Catalysis B: EnvironmentalDocument7 pagesApplied Catalysis B: EnvironmentalAnonymous Wcj4C3jNo ratings yet
- Bioanor 1Document28 pagesBioanor 1Esy Anugerah R KNo ratings yet
- 2022 Cobaloxime MoleculesDocument18 pages2022 Cobaloxime MoleculesLuis SeijasNo ratings yet
- Oxidative Desulfurization of Model Oil IDocument12 pagesOxidative Desulfurization of Model Oil IJorge Rodrigo GranadosNo ratings yet
- 2021, Strategies For Improving Perovskite Photocatalysts Reactivity For OrganicDocument23 pages2021, Strategies For Improving Perovskite Photocatalysts Reactivity For OrganicTRINH HUỲNH NGỌC DIỄMNo ratings yet
- Arora Et Al 2023 Rapid Assembly of Unsymmetrically Linked Bis Heterocycles Via Palladium Domino CatalysisDocument5 pagesArora Et Al 2023 Rapid Assembly of Unsymmetrically Linked Bis Heterocycles Via Palladium Domino Catalysisrizzo.lorenzo94No ratings yet
- JournalDocument8 pagesJournalDwi AdityaNo ratings yet
- Bai 3 Hoa Thien NhienDocument18 pagesBai 3 Hoa Thien NhienĐức Thụ TrầnNo ratings yet
- 2D Bismuth Based LayerdDocument11 pages2D Bismuth Based Layerdhitesh chaudharyNo ratings yet
- Bimetallic Niobium-Based Catalysts Supported On SBA-15 For Hydrodeoxygenation of AnisoleDocument10 pagesBimetallic Niobium-Based Catalysts Supported On SBA-15 For Hydrodeoxygenation of AnisoleMuh Nur Afif LuthfiNo ratings yet
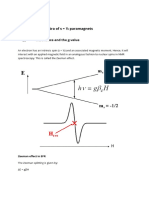
- S > 1⁄2 paramagnets: Zero-field splitting in spin tripletsDocument2 pagesS > 1⁄2 paramagnets: Zero-field splitting in spin tripletsSiddiqui M. M.No ratings yet
- NHCDocument28 pagesNHCSiddiqui M. M.No ratings yet
- Lecture 10Document21 pagesLecture 10Siddiqui M. M.No ratings yet
- 3 EPR Spectroscopy Lectures VI VIII PDFDocument7 pages3 EPR Spectroscopy Lectures VI VIII PDFSiddiqui M. M.No ratings yet
- IsotropicDocument7 pagesIsotropicSiddiqui M. M.No ratings yet
- Inorganics: Mono-And Dinuclear Aluminium Complexes Derived From Biguanide and Carbothiamide LigandsDocument10 pagesInorganics: Mono-And Dinuclear Aluminium Complexes Derived From Biguanide and Carbothiamide LigandsSiddiqui M. M.No ratings yet
- Angew Chem Int Ed - 2022 - Lim - Crystalline Germanium I and Tin I Centered Radical AnionsDocument5 pagesAngew Chem Int Ed - 2022 - Lim - Crystalline Germanium I and Tin I Centered Radical AnionsSiddiqui M. M.No ratings yet
- Valorization of CO: Preparation of 2 Oxazolidinones by Metal Ligand Cooperative Catalysis With SCS Indenediide PD ComplexesDocument9 pagesValorization of CO: Preparation of 2 Oxazolidinones by Metal Ligand Cooperative Catalysis With SCS Indenediide PD ComplexesSiddiqui M. M.No ratings yet
- Cefic-Position-Paper-on-Chemical-Valorisation-of-CO2.pdf# - Text The Chemical Valorisation of CO2, The Physical Utilisation of CO2.Document7 pagesCefic-Position-Paper-on-Chemical-Valorisation-of-CO2.pdf# - Text The Chemical Valorisation of CO2, The Physical Utilisation of CO2.Siddiqui M. M.No ratings yet
- Polymer Molecular Weight and CharacterizationDocument50 pagesPolymer Molecular Weight and CharacterizationSonu PrajapatiNo ratings yet
- Analysis of a rectangular pressure vesselDocument7 pagesAnalysis of a rectangular pressure vesselPressure VesselNo ratings yet
- Technology Update No. 3: SSPC: The Society For Protective CoatingsDocument12 pagesTechnology Update No. 3: SSPC: The Society For Protective CoatingsachusanachuNo ratings yet
- Answer Key Ionic BondingDocument4 pagesAnswer Key Ionic BondingRahmania AviantiNo ratings yet
- 1 ChemistryDocument2 pages1 ChemistryAdik Sharma100% (1)
- Rtu M Tech Thesis FormatDocument6 pagesRtu M Tech Thesis Formataprilscrantonspringfield100% (2)
- ANCARBON Technical Data SheetsDocument24 pagesANCARBON Technical Data SheetsMashiur RahmanNo ratings yet
- Separating Coloured Inks by Paper ChromatographyDocument36 pagesSeparating Coloured Inks by Paper ChromatographyMANSI CHAUDHARYNo ratings yet
- Ep2670803b1 PDFDocument22 pagesEp2670803b1 PDFEnrique EscobarNo ratings yet
- How Many Water Molecules Does It Take To Dissociate HCL?: Doi: 10.1002/chem.201504016Document7 pagesHow Many Water Molecules Does It Take To Dissociate HCL?: Doi: 10.1002/chem.201504016Exlonk Gil PeláezNo ratings yet
- Aking Osmetics: Certificate of AnalysisDocument1 pageAking Osmetics: Certificate of AnalysisAGATHA RIA BUDIYANANo ratings yet
- 1 s2.0 S095006181933380X AmDocument13 pages1 s2.0 S095006181933380X AmvajdazitaNo ratings yet
- International Journal of Biological MacromoleculesDocument9 pagesInternational Journal of Biological MacromoleculesFaizhal Dimas LeksonoNo ratings yet
- A Report On Boiler Feed WaterDocument11 pagesA Report On Boiler Feed WaterAustin UdofiaNo ratings yet
- Mini Project Final ReportDocument37 pagesMini Project Final ReportSamanth Kumar Vasam86% (14)
- A - Review - On - Stability - Studies - of - Pharmaceutical - ProductsDocument9 pagesA - Review - On - Stability - Studies - of - Pharmaceutical - Productsmarco hernandezNo ratings yet
- 15.salt Lead Nitrate 1Document2 pages15.salt Lead Nitrate 1Sarthika GaulkarNo ratings yet
- Biotechnology 1609322314Document100 pagesBiotechnology 1609322314Abdullah SaeedNo ratings yet
- Science Worksheet Grade 7Document2 pagesScience Worksheet Grade 7Ricky KhoasebNo ratings yet
- Ab_Oxidation and Deamidation of Monoclonal Antibody Products- Potential Impact on Stability Biological Activity and EfficacyDocument16 pagesAb_Oxidation and Deamidation of Monoclonal Antibody Products- Potential Impact on Stability Biological Activity and EfficacyCher IshNo ratings yet
- Periodic Table TrendsDocument1 pagePeriodic Table TrendsClick LinkNo ratings yet
- Fosroc Conplast CNI: Constructive SolutionsDocument2 pagesFosroc Conplast CNI: Constructive SolutionsVincent JavateNo ratings yet
- Leather Terms GuideDocument162 pagesLeather Terms GuideAnik AlamNo ratings yet
- Chemistry Block-D: Complex Formation Coordination NumberDocument23 pagesChemistry Block-D: Complex Formation Coordination NumberNurhadi BNo ratings yet
- E Book - Complete Guide of Polycarbonate Fabrication - ExceliteDocument49 pagesE Book - Complete Guide of Polycarbonate Fabrication - ExcelitecpcdbrNo ratings yet
- IRJET-V5I8154-Response Spectrum Modelling Intze Tank PDFDocument6 pagesIRJET-V5I8154-Response Spectrum Modelling Intze Tank PDFRamkumar KumaresanNo ratings yet
- 1112283stock2075 3 17Document5 pages1112283stock2075 3 17cds nepalNo ratings yet
- CBC COVID19 Product List 3 - 25 - 2020 PDFDocument15 pagesCBC COVID19 Product List 3 - 25 - 2020 PDFIgnaMarounNo ratings yet
- VPF-10-50 - Water based co-polyester for excellent adhesion on polyester filmsDocument6 pagesVPF-10-50 - Water based co-polyester for excellent adhesion on polyester filmsatharv swaroopNo ratings yet
- (Solved) Hydration of 1-Hexene 1-Hexene To 2 Hexanol Equation Write The Equation of The Reaction - Course HeroDocument2 pages(Solved) Hydration of 1-Hexene 1-Hexene To 2 Hexanol Equation Write The Equation of The Reaction - Course HeroJdkrkejNo ratings yet