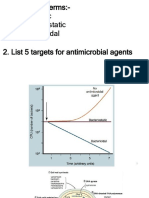
You might also like
- Enzymes: Biochemistry, Biotechnology, Clinical ChemistryFrom EverandEnzymes: Biochemistry, Biotechnology, Clinical ChemistryRating: 4 out of 5 stars4/5 (8)
- Idowu 2020Document9 pagesIdowu 2020Sherwan HusseinNo ratings yet
- 12 2017 Beta-Lactamases A Focus Current ChallengesDocument15 pages12 2017 Beta-Lactamases A Focus Current ChallengesAlisonNo ratings yet
- Discovery and Development of CephalosporinsDocument7 pagesDiscovery and Development of Cephalosporinsolivia523No ratings yet
- β-Lactamases and β-Lactamase Inhibitors in the 21st CenturyDocument29 pagesβ-Lactamases and β-Lactamase Inhibitors in the 21st CenturyMarisol Estefany FarfanNo ratings yet
- Barton EllaDocument16 pagesBarton Ellamurti_fatiyaNo ratings yet
- Fmicb-13-810403 Klebsiella 2022 Colistino Pero Solo PE PARA MEMBRANASDocument18 pagesFmicb-13-810403 Klebsiella 2022 Colistino Pero Solo PE PARA MEMBRANASmbalimentoslspdNo ratings yet
- Biochimica Et Biophysica Acta: Panagiota K. Kyriakou, Bie Ekblad, Per Eugen Kristiansen, Yiannis N. KaznessisDocument12 pagesBiochimica Et Biophysica Acta: Panagiota K. Kyriakou, Bie Ekblad, Per Eugen Kristiansen, Yiannis N. KaznessisAyu Asriningati PNo ratings yet
- Pharmd - 3Y - 3.5 - Medicinal ChemistryDocument41 pagesPharmd - 3Y - 3.5 - Medicinal ChemistrySyed Ali ShahNo ratings yet
- HHS Public Access: Efficacy of Ampicillin Against Methicillin-ResistantDocument18 pagesHHS Public Access: Efficacy of Ampicillin Against Methicillin-ResistantIraNo ratings yet
- Articulo 3Document5 pagesArticulo 3Karen CaizaNo ratings yet
- Wall Bacteria Cell-1Document15 pagesWall Bacteria Cell-1Richard MoralesNo ratings yet
- Antimicrobial Peptide Structure and MechDocument15 pagesAntimicrobial Peptide Structure and MechmanelmanoNo ratings yet
- Beta LactamideDocument23 pagesBeta LactamideDana GeorgianaNo ratings yet
- MX Resist Gram+Document8 pagesMX Resist Gram+Yorvi hendersson Ilasaca gaonaNo ratings yet
- Mechanisms of Antibiotic Action, Class PPT IIDocument13 pagesMechanisms of Antibiotic Action, Class PPT IILarry TongNo ratings yet
- Antibiotics IIDocument12 pagesAntibiotics IIMuhammad AliNo ratings yet
- Resistensi Dan Cara Kerja AntiboticsDocument6 pagesResistensi Dan Cara Kerja AntiboticsChristiadi karlin12No ratings yet
- Beta-Lactam & Other Cell Wall-& Membrane-Active AntibioticsDocument43 pagesBeta-Lactam & Other Cell Wall-& Membrane-Active AntibioticsTutde SedanaNo ratings yet
- Beta 2 NotesDocument14 pagesBeta 2 Notesdita anisya putriNo ratings yet
- Degradation of: ββ-lactam antibioticsDocument12 pagesDegradation of: ββ-lactam antibioticsMuhammad Azam TahirNo ratings yet
- Peniciilins and Beta LactamsDocument134 pagesPeniciilins and Beta LactamsMelissa TisadoNo ratings yet
- Beta-Lactam Antibiotics Kul Nov 2014Document44 pagesBeta-Lactam Antibiotics Kul Nov 2014Sahala Louis AlexanderNo ratings yet
- Bacterial Antibiotics: Mechanisms of ResistanceDocument10 pagesBacterial Antibiotics: Mechanisms of ResistanceAnnisa YohanesNo ratings yet
- Beta-Lactam Antibiotics: Mechanisms of Action and Resistance and Adverse EffectsDocument13 pagesBeta-Lactam Antibiotics: Mechanisms of Action and Resistance and Adverse EffectsellaNo ratings yet
- Beta LactamsDocument22 pagesBeta LactamsRoberto PerezNo ratings yet
- Spectrum 01198-23Document16 pagesSpectrum 01198-23Robert StryjakNo ratings yet
- Mechanisms of Antibiotic Resistance: DR T. Aswani Ndonga MSC Tid I April 2010Document25 pagesMechanisms of Antibiotic Resistance: DR T. Aswani Ndonga MSC Tid I April 2010GAMING MONKEYNo ratings yet
- Optimization of 4 Aminoquinoline/Clotrimazole-Based Hybrid Antimalarials: Further Structure Activity Relationships, in Vivo Studies, and Preliminary Toxicity Pro FilingDocument20 pagesOptimization of 4 Aminoquinoline/Clotrimazole-Based Hybrid Antimalarials: Further Structure Activity Relationships, in Vivo Studies, and Preliminary Toxicity Pro FilinganacarolinacsousaNo ratings yet
- Antibiotic ClassificationDocument10 pagesAntibiotic ClassificationRipka_uliNo ratings yet
- Artigo Iluska - Lin Et Al 2020Document10 pagesArtigo Iluska - Lin Et Al 2020larissa dias freitasNo ratings yet
- Computational Calculations of Molecular Properties and Molecular Docking of New and Reference Cephalosporins on Penicillin Binding Proteins and Various β-LactamasesDocument14 pagesComputational Calculations of Molecular Properties and Molecular Docking of New and Reference Cephalosporins on Penicillin Binding Proteins and Various β-LactamasesSabrina JonesNo ratings yet
- Fan2018 PDFDocument27 pagesFan2018 PDFRev EngNo ratings yet
- Abiola Muhammad, ADEOSUN Department of Biochemistry, Lead City University, IbadanDocument37 pagesAbiola Muhammad, ADEOSUN Department of Biochemistry, Lead City University, IbadanKhushi PatelNo ratings yet
- Can Penicillins and Other B-Lactam Antibiotics Be Used To Treat Tuberculosis?Document5 pagesCan Penicillins and Other B-Lactam Antibiotics Be Used To Treat Tuberculosis?EknoorNo ratings yet
- Cardinal Requirements Namely:: Capable, in Small Concentrations, of Inhibiting The Life Processes of Micro-Organisms.'Document20 pagesCardinal Requirements Namely:: Capable, in Small Concentrations, of Inhibiting The Life Processes of Micro-Organisms.'SalmanNo ratings yet
- JMC2013 A Baulmannii Siderophoric AntibacterialDocument9 pagesJMC2013 A Baulmannii Siderophoric AntibacterialVincent GeruszNo ratings yet
- Microbial PathogenesisDocument9 pagesMicrobial Pathogenesislulipampin012No ratings yet
- Abodakpi 2019 PKPDDocument17 pagesAbodakpi 2019 PKPDCarlos Espinoza CobeñasNo ratings yet
- ART Action and Resistance Mechanisms of Antibiotics A Guide For CliniciansDocument16 pagesART Action and Resistance Mechanisms of Antibiotics A Guide For CliniciansHECTORIBZAN ACERO SANDOVALNo ratings yet
- Antibiotics 09 00553 PDFDocument21 pagesAntibiotics 09 00553 PDFGepetto ArtsNo ratings yet
- Antibiotic Resistance PDFDocument6 pagesAntibiotic Resistance PDFGea MarieNo ratings yet
- Please Use Endnote Program To Organize Your ReferencesDocument25 pagesPlease Use Endnote Program To Organize Your ReferencesMy PlanNo ratings yet
- IJAA - CRAb - Jaidane 2018Document4 pagesIJAA - CRAb - Jaidane 2018Nadia JaidaneNo ratings yet
- Printed in Great Britain. All Rights Reserved, ©: Biotech. Adv. Vol. 9, Pp. 217-240,1991Document24 pagesPrinted in Great Britain. All Rights Reserved, ©: Biotech. Adv. Vol. 9, Pp. 217-240,1991sebastian floresNo ratings yet
- Beta-Lactam Antibiotics & Other Inhibitors of Cell WallDocument71 pagesBeta-Lactam Antibiotics & Other Inhibitors of Cell WallAlvin LaurenceNo ratings yet
- Cell Wall Inhibitors: Beta-Lactams: PenicillinsDocument12 pagesCell Wall Inhibitors: Beta-Lactams: Penicillinsمحمد بن صالحNo ratings yet
- Discovery of PenicillinDocument25 pagesDiscovery of PenicillinBhagyashree BachhavNo ratings yet
- Mechanisms of Antibiotic Resistance: DR T. Aswani Ndonga MSC Tid I April 2010Document25 pagesMechanisms of Antibiotic Resistance: DR T. Aswani Ndonga MSC Tid I April 2010sushantk862No ratings yet
- Pharmacology AT GLIMPSE PDFDocument252 pagesPharmacology AT GLIMPSE PDFBiswarup Das100% (1)
- PenicillinsDocument15 pagesPenicillinsFaisalNo ratings yet
- Mvab 102Document14 pagesMvab 102DCPNo ratings yet
- AntibioticosDocument11 pagesAntibioticosdroswaldo88No ratings yet
- JMC2014 Boronic Acid B-Lactam InhibitorDocument10 pagesJMC2014 Boronic Acid B-Lactam InhibitorVincent GeruszNo ratings yet
- Beta-Lactam Antibiotics Will Bind To Serine Proteases (TranspeptidaseDocument4 pagesBeta-Lactam Antibiotics Will Bind To Serine Proteases (TranspeptidaseJames RussellNo ratings yet
- Antibiotics IntroductiontoClassificationDocument16 pagesAntibiotics IntroductiontoClassificationFarida CitraNo ratings yet
- Ijerph 20 01900 v2Document11 pagesIjerph 20 01900 v2edisonballaNo ratings yet
- Cell Wall Inhibitors: Zarqa University Pharmacy School Clinical Pharmacy and Therapeutics Department Pharmacology IIIDocument45 pagesCell Wall Inhibitors: Zarqa University Pharmacy School Clinical Pharmacy and Therapeutics Department Pharmacology IIIAla'a AL AqrabawyNo ratings yet
- 1 s2.0 S1047847715000684 Main PDFDocument11 pages1 s2.0 S1047847715000684 Main PDFAsa Étudier La-DienNo ratings yet
- Tetrahedron2005,61,11850 Muraymycin U MalagaDocument16 pagesTetrahedron2005,61,11850 Muraymycin U MalagaVincent GeruszNo ratings yet
- BMCL2002,12,2341 Muraymycin WyethDocument4 pagesBMCL2002,12,2341 Muraymycin WyethVincent GeruszNo ratings yet
- TL1996 MeyersDocument2 pagesTL1996 MeyersVincent GeruszNo ratings yet
- JMC2012 DNA Helicase Inhibitors MicrobiotixDocument13 pagesJMC2012 DNA Helicase Inhibitors MicrobiotixVincent GeruszNo ratings yet
- JMC2010,53,5502 Gossypol-APADocument9 pagesJMC2010,53,5502 Gossypol-APAVincent GeruszNo ratings yet
- BMCL2003,13,3345 Muraymycin WyethDocument6 pagesBMCL2003,13,3345 Muraymycin WyethVincent GeruszNo ratings yet
- TetrahedronAsymm2008,19,397 MraY Inhibitor CNRSDocument4 pagesTetrahedronAsymm2008,19,397 MraY Inhibitor CNRSVincent GeruszNo ratings yet
- JMC2003 ReedDocument6 pagesJMC2003 ReedVincent GeruszNo ratings yet
- ACSMCL 2013 Acid Sulfonamide ABT737 NovartisDocument5 pagesACSMCL 2013 Acid Sulfonamide ABT737 NovartisVincent GeruszNo ratings yet
- JMC2014 InhA InhibitorsDocument13 pagesJMC2014 InhA InhibitorsVincent GeruszNo ratings yet
- Chem Bio 2004 PellecchiaDocument7 pagesChem Bio 2004 PellecchiaVincent GeruszNo ratings yet
- JMC2013 SAR Benzimidazole TB FTSZDocument15 pagesJMC2013 SAR Benzimidazole TB FTSZVincent GeruszNo ratings yet
- Bio Med Chem 2005 GroundwaterDocument10 pagesBio Med Chem 2005 GroundwaterVincent GeruszNo ratings yet
- Jmc2013 Inha AzDocument10 pagesJmc2013 Inha AzVincent GeruszNo ratings yet
- ACSMedChemLett2010 TherisodDocument4 pagesACSMedChemLett2010 TherisodVincent GeruszNo ratings yet
- JMC2013 TB IndolecarboxamideDocument11 pagesJMC2013 TB IndolecarboxamideVincent GeruszNo ratings yet
- ACSMedChemLett 2013 TB ImidazopyridinesDocument5 pagesACSMedChemLett 2013 TB ImidazopyridinesVincent GeruszNo ratings yet
- JMolBiol1999 X-Ray FBA E Coli Et PGHDocument12 pagesJMolBiol1999 X-Ray FBA E Coli Et PGHVincent GeruszNo ratings yet
- JMC2013 DFG Kinase InhibitorsDocument15 pagesJMC2013 DFG Kinase InhibitorsVincent GeruszNo ratings yet
- JMC2013 Azaindoles TB VivoDocument8 pagesJMC2013 Azaindoles TB VivoVincent GeruszNo ratings yet
- JBC 2002 FBA Vs TBADocument7 pagesJBC 2002 FBA Vs TBAVincent GeruszNo ratings yet
- PlosOne 2012 yycFGHIDocument11 pagesPlosOne 2012 yycFGHIVincent GeruszNo ratings yet
- JMC2010 TherisodDocument7 pagesJMC2010 TherisodVincent GeruszNo ratings yet
- JMC2013 Indolecarboxamide anti-TB KozikowskiDocument11 pagesJMC2013 Indolecarboxamide anti-TB KozikowskiVincent GeruszNo ratings yet
- ACSMedChemLett2013 Hemolytic Conc Gram Neg Integrity MomentDocument5 pagesACSMedChemLett2013 Hemolytic Conc Gram Neg Integrity MomentVincent GeruszNo ratings yet
- BMCL2007 YycG Inhibitors Kinki UniversityDocument4 pagesBMCL2007 YycG Inhibitors Kinki UniversityVincent GeruszNo ratings yet
- JMC2013 A Baulmannii Siderophoric AntibacterialDocument9 pagesJMC2013 A Baulmannii Siderophoric AntibacterialVincent GeruszNo ratings yet
- JMC2013 Cyclin Dependent Kinase InhibitorDocument15 pagesJMC2013 Cyclin Dependent Kinase InhibitorVincent GeruszNo ratings yet
- BMCMicrobiology 2006 YycG Inhibitors ShangaiDocument18 pagesBMCMicrobiology 2006 YycG Inhibitors ShangaiVincent GeruszNo ratings yet
- Ann N Y Acad Sci. 2012 Mechanism of Dapto Resistance Involving Change in yycGFDocument20 pagesAnn N Y Acad Sci. 2012 Mechanism of Dapto Resistance Involving Change in yycGFVincent GeruszNo ratings yet
- О-Acetylation of 4-Hydroxybenzoic Acid with Acetic AnhydrideDocument6 pagesО-Acetylation of 4-Hydroxybenzoic Acid with Acetic AnhydrideRichaNo ratings yet
- BR Matacryl WS - GBDocument4 pagesBR Matacryl WS - GBNevena DelibasicNo ratings yet
- 15 - Mangla Joshi PDFDocument22 pages15 - Mangla Joshi PDFSHIKHA SINGHNo ratings yet
- Opeartion ManualDocument36 pagesOpeartion ManualZachi UkiNo ratings yet
- Fixation & FixativesDocument64 pagesFixation & FixativesMadhura Shekatkar100% (1)
- VItamin K2 & Macular DegenerationDocument5 pagesVItamin K2 & Macular DegenerationfereyNo ratings yet
- Angew Chem Int Ed - 2020 - Sarkar - A Neutral Three Membered 2 Aromatic Disilaborirane and The Unique Conversion Into ADocument5 pagesAngew Chem Int Ed - 2020 - Sarkar - A Neutral Three Membered 2 Aromatic Disilaborirane and The Unique Conversion Into ATutu CaiNo ratings yet
- General Chemistry 2 - Grade 11Document16 pagesGeneral Chemistry 2 - Grade 11Adoneza Gwyn AgustinNo ratings yet
- Ul 746B 2011 PDFDocument56 pagesUl 746B 2011 PDFShibu1992No ratings yet
- Kjeldahl MethodDocument3 pagesKjeldahl MethodNMLNo ratings yet
- Leffet Inhibiteur Deugenol Sur La CorrosDocument11 pagesLeffet Inhibiteur Deugenol Sur La CorrosFadwa AsriNo ratings yet
- Module-3 (Part-I)Document218 pagesModule-3 (Part-I)Prajay GNo ratings yet
- Ge Ni U S: Chemical ReactionsDocument25 pagesGe Ni U S: Chemical ReactionsRouda AljNo ratings yet
- Novel Hydrotreating Technology For Production of Green Diesel - Haldor TopsoeDocument21 pagesNovel Hydrotreating Technology For Production of Green Diesel - Haldor Topsoebalarie100% (2)
- Rate LawDocument3 pagesRate Lawnadia sykesNo ratings yet
- Chemistry Class 10 Chapter 10Document15 pagesChemistry Class 10 Chapter 10Rahim BakhshNo ratings yet
- 4 - CHAPTER With NumericalsDocument356 pages4 - CHAPTER With NumericalsSindhuja PachikaNo ratings yet
- Nanoemulsions of Essential Oils To Improve Solubility, Stability and Permeability: A ReviewDocument19 pagesNanoemulsions of Essential Oils To Improve Solubility, Stability and Permeability: A ReviewHector LeónNo ratings yet
- Document 32Document17 pagesDocument 32rik.sengupta.08No ratings yet
- Components of Coal AshDocument4 pagesComponents of Coal AshDulguun BayNo ratings yet
- Moores Test and Barfoeds TestDocument3 pagesMoores Test and Barfoeds TestFrancis CaloNo ratings yet
- Hydroprocessing: Hydrocracking & HydrotreatingDocument45 pagesHydroprocessing: Hydrocracking & HydrotreatingRobin ZwartNo ratings yet
- Chromtaography - GCDocument18 pagesChromtaography - GCnajdat alzaatraNo ratings yet
- A Foot Ahead of The Pink SnowDocument15 pagesA Foot Ahead of The Pink SnowGreater Potential TutoringNo ratings yet
- Colorants and Auxiliaries Vol 2Document455 pagesColorants and Auxiliaries Vol 2Putri Mayangsari100% (11)
- SCOPE 21 - The Major Biogeochemical Cycles and Their InteractionsDocument205 pagesSCOPE 21 - The Major Biogeochemical Cycles and Their InteractionsAlina JalbaNo ratings yet
- S17 2170501 CreDocument3 pagesS17 2170501 CremorganNo ratings yet
- Chap 1Document53 pagesChap 1patilamardip0078122No ratings yet
- O Level Past Papers 5070 - s16 - QP - 41Document16 pagesO Level Past Papers 5070 - s16 - QP - 41Hamad SaeidNo ratings yet
- Coq 004DP44Document2 pagesCoq 004DP44m100% (2)