You might also like
- Clinical Applied Anatomy in Wound CareDocument18 pagesClinical Applied Anatomy in Wound CareKlinik Komuniti Perwira100% (2)
- Lesson 5: Tabata TrainingDocument31 pagesLesson 5: Tabata TrainingAshley Keith RamiloNo ratings yet
- Car T Celll TherapyDocument45 pagesCar T Celll TherapyNiraj Singh100% (2)
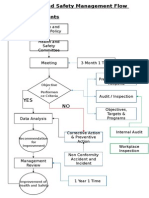
- Health and Safety FlowDocument6 pagesHealth and Safety Flowzaki0304No ratings yet
- Fast Facts: CAR T-Cell Therapy: Insight into current and future applicationsFrom EverandFast Facts: CAR T-Cell Therapy: Insight into current and future applicationsNo ratings yet
- Armstrong'S Handbook Of: Human Resource ManagementDocument69 pagesArmstrong'S Handbook Of: Human Resource ManagementNaina lalwaniNo ratings yet
- Global CAR T Cell Therapy Market and Clinical Trials Insight 2022Document5 pagesGlobal CAR T Cell Therapy Market and Clinical Trials Insight 2022Neeraj ChawlaNo ratings yet
- Set 5Document18 pagesSet 5Dick Morgan FerrerNo ratings yet
- TFN ReviewerDocument18 pagesTFN Reviewerkiki park100% (1)
- In Vivo Fate and Activity of Second - Versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin's LymphomasDocument11 pagesIn Vivo Fate and Activity of Second - Versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin's LymphomasNestor AndresNo ratings yet
- Lang 2015Document5 pagesLang 2015Diego SantoroNo ratings yet
- Chimeric Antigen Receptor Engineered Human Gamma DDocument12 pagesChimeric Antigen Receptor Engineered Human Gamma Dedwin tjandraNo ratings yet
- 5 I New Car Constructs and Targets in Clinical TrialsDocument4 pages5 I New Car Constructs and Targets in Clinical TrialsFatima Al SayyedNo ratings yet
- Car-T Cells TherapyDocument7 pagesCar-T Cells TherapydrbrevathiNo ratings yet
- CD19 Followed by Allogenic Stem CellDocument2 pagesCD19 Followed by Allogenic Stem CelljohnNo ratings yet
- Preclinical Development and Evaluation of Nanobody-Based CD70-specific CAR T Cells For The Treatment of Acute Myeloid LeukemiaDocument16 pagesPreclinical Development and Evaluation of Nanobody-Based CD70-specific CAR T Cells For The Treatment of Acute Myeloid LeukemiaMilan JovicNo ratings yet
- Multiplex Immunohistochemistry Defines The Tumor Immune Microenvironment and Immunotherapeutic Outcome in CLDN18.2-positive Gastric CancerDocument15 pagesMultiplex Immunohistochemistry Defines The Tumor Immune Microenvironment and Immunotherapeutic Outcome in CLDN18.2-positive Gastric CancerDr Meenakshi ParwaniNo ratings yet
- Results of ARI-0001 CART19 Cells in Patients WithDocument7 pagesResults of ARI-0001 CART19 Cells in Patients WithJuhi VermaNo ratings yet
- Chimeric Antigen Receptor T Cell Therapy For Non-Hodgkin LymphomaDocument7 pagesChimeric Antigen Receptor T Cell Therapy For Non-Hodgkin Lymphomagatorfan786No ratings yet
- Car-T Cell 1 (7760)Document10 pagesCar-T Cell 1 (7760)Saradha PellatiNo ratings yet
- 1 - Relmacabtagene Autoleucel (Relma-Cel) CD19 CAR-T Therapy For Adults With Heavily Pretreated Relapsed or Refractory Large B-Cell Lymphoma in ChinaDocument13 pages1 - Relmacabtagene Autoleucel (Relma-Cel) CD19 CAR-T Therapy For Adults With Heavily Pretreated Relapsed or Refractory Large B-Cell Lymphoma in ChinaFaras ArinalNo ratings yet
- 1 s2.0 S0006497121069597 MainDocument13 pages1 s2.0 S0006497121069597 Mainf2xt4fj858No ratings yet
- Recent Advances in CAR-T Cell Engineering: Review Open AccessDocument19 pagesRecent Advances in CAR-T Cell Engineering: Review Open AccessAtrocitus RedNo ratings yet
- Humanized Anti-Cd26 Monoclonal Antibody As A Treatment For Malignant MesotheliomatumorsDocument11 pagesHumanized Anti-Cd26 Monoclonal Antibody As A Treatment For Malignant MesotheliomatumorsRezultate DMD ElytisNo ratings yet
- A Game Changer in Cancer TreatmentDocument11 pagesA Game Changer in Cancer TreatmentŞeyma YılmazNo ratings yet
- MA - Evidence of Long Lasting Anti CD19 Activity of Engrafted CD19 Chimeric Antigen - DATADocument8 pagesMA - Evidence of Long Lasting Anti CD19 Activity of Engrafted CD19 Chimeric Antigen - DATAdaniela santurioNo ratings yet
- Car-T Cell Receptors (7758)Document8 pagesCar-T Cell Receptors (7758)Saradha PellatiNo ratings yet
- Research Article: Shimu Luo, Yanghang Ou, Tingjin Zheng, Huihui Jiang, Yibo Wu, Jiangman Zhao, and Zhishan ZhangDocument9 pagesResearch Article: Shimu Luo, Yanghang Ou, Tingjin Zheng, Huihui Jiang, Yibo Wu, Jiangman Zhao, and Zhishan ZhangcecilliacynthiaNo ratings yet
- Mto 201615Document7 pagesMto 201615Tim TiemannNo ratings yet
- Immunology - 2008 - Montcuquet - Regulatory T Cell Expansion and Function Do Not Account For The Impaired Alloreactivity ofDocument11 pagesImmunology - 2008 - Montcuquet - Regulatory T Cell Expansion and Function Do Not Account For The Impaired Alloreactivity ofciara babyNo ratings yet
- Li 2020Document13 pagesLi 2020Maria Carolina CoutoNo ratings yet
- Car-T Cell Therapy - Final DraftDocument50 pagesCar-T Cell Therapy - Final DraftFatina HatoumNo ratings yet
- Journal Pre-Proof: Seminars in HematologyDocument23 pagesJournal Pre-Proof: Seminars in HematologyAtrocitus RedNo ratings yet
- (03241750 - Acta Medica Bulgarica) Extensive Proliferation of Cd4+ Lymphocyte by Both Phytohaemagglutinin A and Anti-Cd2 - Cd3 - Cd28 Macsibeads PDFDocument4 pages(03241750 - Acta Medica Bulgarica) Extensive Proliferation of Cd4+ Lymphocyte by Both Phytohaemagglutinin A and Anti-Cd2 - Cd3 - Cd28 Macsibeads PDFTeodorNo ratings yet
- 1 s2.0 S2372770520300954 MainDocument10 pages1 s2.0 S2372770520300954 MainJuhi VermaNo ratings yet
- Genetic Modi Fication of Tumor-In Filtrating Lymphocytes Via Retroviral TransductionDocument11 pagesGenetic Modi Fication of Tumor-In Filtrating Lymphocytes Via Retroviral TransductiondianaNo ratings yet
- 2021 SupimonDocument14 pages2021 SupimonMutita JunkingNo ratings yet
- Nej Mo A 2308917Document14 pagesNej Mo A 2308917mnf6bb2tckNo ratings yet
- Intratumoral IL-12 Delivery Empowers CAR-T Cell Immunotherapy in A Pre-Clinical Model of GlioblastomaDocument11 pagesIntratumoral IL-12 Delivery Empowers CAR-T Cell Immunotherapy in A Pre-Clinical Model of GlioblastomaZEKUN ZHAONo ratings yet
- 1099 Full PDFDocument6 pages1099 Full PDFWahyu WijayantoNo ratings yet
- Expression of High Amounts of The CD117 Molecule in A Case of B-Cell Non-Hodgkin's Lymphoma Carrying The T (14:18) TranslocationDocument4 pagesExpression of High Amounts of The CD117 Molecule in A Case of B-Cell Non-Hodgkin's Lymphoma Carrying The T (14:18) Translocationawdafeagega2r3No ratings yet
- ASH Hematology Review Series - CAR-T Porter Sub 8-5-21Document63 pagesASH Hematology Review Series - CAR-T Porter Sub 8-5-21Иван НегарэNo ratings yet
- CRISPR Takes The Front Seat in CART Cell DevelopmentDocument12 pagesCRISPR Takes The Front Seat in CART Cell DevelopmentThiên Kim LêNo ratings yet
- Intrathyroid Injection of Dexamethasone Inhibits Th2 Cells in Graves' DiseaseDocument8 pagesIntrathyroid Injection of Dexamethasone Inhibits Th2 Cells in Graves' DiseaselolaNo ratings yet
- 1 s2.0 S0168365922005387 MainDocument10 pages1 s2.0 S0168365922005387 Maindylan.sjdaiNo ratings yet
- Stromal CD4+ and CD8+ T Cells in Human Breast Carcinomas. Its Correlation With Chemokine Mig/Cxcl9Document5 pagesStromal CD4+ and CD8+ T Cells in Human Breast Carcinomas. Its Correlation With Chemokine Mig/Cxcl9Uum SudrajatNo ratings yet
- Clinical Manufacturing of CAR T Cells FoundationDocument7 pagesClinical Manufacturing of CAR T Cells FoundationMinh Trần ThịNo ratings yet
- Kobbe 1998Document4 pagesKobbe 1998dad dzd adaNo ratings yet
- +CD103+CD8+ T Cells in Human High-Grade: Transcriptional Activity and Stability of CD39 Endometrial CancerDocument18 pages+CD103+CD8+ T Cells in Human High-Grade: Transcriptional Activity and Stability of CD39 Endometrial CancerSeda KNo ratings yet
- 2015-CDD-Reciprocal Positive Regulation Between Cx26 and PI3K:Akt Pathway Confers Acquired Gefitinib Resistance in NSCLC Cells Via GJIC-independent Induction of EMTDocument12 pages2015-CDD-Reciprocal Positive Regulation Between Cx26 and PI3K:Akt Pathway Confers Acquired Gefitinib Resistance in NSCLC Cells Via GJIC-independent Induction of EMTzhe zhNo ratings yet
- ChAd mRNA Solid TumorsDocument39 pagesChAd mRNA Solid TumorsMartin PommeretNo ratings yet
- 2022 Article 4489Document30 pages2022 Article 4489wiwiNo ratings yet
- 1 s2.0 S2372770519300737 MainDocument9 pages1 s2.0 S2372770519300737 MainJuhi VermaNo ratings yet
- CAR T改造大法 PDFDocument16 pagesCAR T改造大法 PDFTianliang GuoNo ratings yet
- Gpc3 Generate de Celule Dendritice 2010Document11 pagesGpc3 Generate de Celule Dendritice 2010grigmihNo ratings yet
- The Response To Lymphodepletion Impacts PFS in Patients With Aggressive Non-Hodgkin Lymphoma Treated With CD19 CAR T CellsDocument12 pagesThe Response To Lymphodepletion Impacts PFS in Patients With Aggressive Non-Hodgkin Lymphoma Treated With CD19 CAR T CellsDemi D. ArciniegaNo ratings yet
- Cas 14835Document12 pagesCas 14835Sediki ZakariaNo ratings yet
- CAR T Therapy Beyond Cancer: The Evolution of A Living DrugDocument14 pagesCAR T Therapy Beyond Cancer: The Evolution of A Living DrugCinta DíezNo ratings yet
- Engineering The Next-Generation of CAR T-Cells With CRISPR-Cas9 Gene EditingDocument13 pagesEngineering The Next-Generation of CAR T-Cells With CRISPR-Cas9 Gene Editingmoussodjirebecca9No ratings yet
- Off-The-Shelf' Allogeneic CAR T Cells: Development and ChallengesDocument15 pagesOff-The-Shelf' Allogeneic CAR T Cells: Development and ChallengesMatheus ArengheriNo ratings yet
- Bridging Chemotherapy ALLDocument4 pagesBridging Chemotherapy ALL49qmzqdpndNo ratings yet
- Prognostic Relevance of The Expression of TDT and CD7 in 335 Cases of Acute Myeloid LeukemiaDocument8 pagesPrognostic Relevance of The Expression of TDT and CD7 in 335 Cases of Acute Myeloid LeukemiaAnonymous iqoU1mtNo ratings yet
- FCVM 10 1090103Document9 pagesFCVM 10 1090103lipu1982No ratings yet
- Foy Et Al 2023 NatureDocument32 pagesFoy Et Al 2023 Natureneosquall89No ratings yet
- Immune Function in Children Under Chemotherapy For Standard Risk Acute Lymphoblastic Leukaemia - A Prospective Study of 20 Paediatric PatientsDocument11 pagesImmune Function in Children Under Chemotherapy For Standard Risk Acute Lymphoblastic Leukaemia - A Prospective Study of 20 Paediatric PatientsJOSE PORTILLONo ratings yet
- Pnas 2304319120 SappDocument15 pagesPnas 2304319120 SappfilymascoloNo ratings yet
- Automated Manufacture of Autologous CD19 CAR-T CelDocument13 pagesAutomated Manufacture of Autologous CD19 CAR-T CelJuhi VermaNo ratings yet
- Results of ARI-0001 CART19 Cells in Patients WithDocument7 pagesResults of ARI-0001 CART19 Cells in Patients WithJuhi VermaNo ratings yet
- De Lima 20Document14 pagesDe Lima 20Juhi VermaNo ratings yet
- PRG 1801Document2 pagesPRG 1801Juhi VermaNo ratings yet
- Next Gen CARDocument11 pagesNext Gen CARJuhi VermaNo ratings yet
- Cells: Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling ThresholdDocument17 pagesCells: Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling ThresholdJuhi VermaNo ratings yet
- 1 s2.0 S2372770520300954 MainDocument10 pages1 s2.0 S2372770520300954 MainJuhi VermaNo ratings yet
- 1 s2.0 S0952791521000893 MainDocument9 pages1 s2.0 S0952791521000893 MainJuhi VermaNo ratings yet
- 1 s2.0 S2372770519300737 MainDocument9 pages1 s2.0 S2372770519300737 MainJuhi VermaNo ratings yet
- 1 Script200302090903033030Document9 pages1 Script200302090903033030gever29816No ratings yet
- Gulayan Sa PaaralanDocument5 pagesGulayan Sa PaaralanLani DolleroNo ratings yet
- Ebook Ebook PDF Personal Stress Management From Surviving To Thriving PDFDocument41 pagesEbook Ebook PDF Personal Stress Management From Surviving To Thriving PDFtina.cousins322100% (37)
- RLE-level-2-Learning-packet-2-Blue-week-2 (Sanaani, Nur-Fatima, M.)Document29 pagesRLE-level-2-Learning-packet-2-Blue-week-2 (Sanaani, Nur-Fatima, M.)Nur SanaaniNo ratings yet
- EWC 661 English For Report Writing: Prepared By: NO Name Student IdDocument7 pagesEWC 661 English For Report Writing: Prepared By: NO Name Student IdAthirah Dinata100% (1)
- Pretibial LacsDocument8 pagesPretibial LacsMiguel JohnsonNo ratings yet
- LP 20 Health Q3Document8 pagesLP 20 Health Q3Hazel Rubas SamsonNo ratings yet
- Food AdjunctDocument15 pagesFood AdjunctRoby martinus bayaNo ratings yet
- Application of Gis in Health Informatics ResearchDocument6 pagesApplication of Gis in Health Informatics ResearchRaju KoiralaNo ratings yet
- The Role of Health-Related Behaviors in The Socioeconomic Disparities in Oral HealthDocument6 pagesThe Role of Health-Related Behaviors in The Socioeconomic Disparities in Oral HealthStephanyNo ratings yet
- A Client With Cushing's Syndrome: Nursing Care PlanDocument1 pageA Client With Cushing's Syndrome: Nursing Care PlanJulius Caesar ColladoNo ratings yet
- Curahealth Hospitals Completes The Purchase of Specialty Hospital of JacksonvilleDocument2 pagesCurahealth Hospitals Completes The Purchase of Specialty Hospital of JacksonvillePR.comNo ratings yet
- Introduction To Soya BeanDocument6 pagesIntroduction To Soya BeanMisbah FatimahNo ratings yet
- English For Academic Purposes: Task 2: Oral Presentation (5%)Document11 pagesEnglish For Academic Purposes: Task 2: Oral Presentation (5%)preyangkaNo ratings yet
- Autism Poster Presentation 643Document1 pageAutism Poster Presentation 643api-293219537No ratings yet
- 02 Nutrition in Lifecycle - 2019Document47 pages02 Nutrition in Lifecycle - 2019Meilina Putri100% (1)
- Mental Health LetterDocument2 pagesMental Health LetterBethanyNo ratings yet
- CH 06Document7 pagesCH 06john mwangiNo ratings yet
- Annotated BibliographyDocument20 pagesAnnotated Bibliographyapi-347153077No ratings yet
- R08 HE3C32 TIE PLN HS 0004 Emergency Response PlanDocument27 pagesR08 HE3C32 TIE PLN HS 0004 Emergency Response PlanHafiz ZameerNo ratings yet
- Revision Sistematica Preparacion A La Practica Dental 2018Document9 pagesRevision Sistematica Preparacion A La Practica Dental 2018Chris MartinNo ratings yet
- Opening: (EN-ES-EI-AI-DI) (EN-ES-EI-AI-DI)Document4 pagesOpening: (EN-ES-EI-AI-DI) (EN-ES-EI-AI-DI)Trisna meyanaNo ratings yet
- Promotive and Preventive Health Aspect in CHN & Facts On Healthcare in PHDocument2 pagesPromotive and Preventive Health Aspect in CHN & Facts On Healthcare in PH2A - Nicole Marrie HonradoNo ratings yet
- Pathogen Reduction/Hazard Analysis Critical Control Point (HACCP) Systems Final Rule 9 CFR Part 417Document16 pagesPathogen Reduction/Hazard Analysis Critical Control Point (HACCP) Systems Final Rule 9 CFR Part 417Mili MartinezNo ratings yet