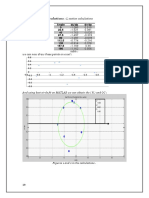
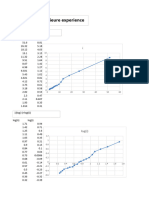
You might also like
- The Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionFrom EverandThe Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionNo ratings yet
- Chee4362 HWDocument8 pagesChee4362 HWHa Eun KimNo ratings yet
- R0-Design Calculation Report of Loading FrameDocument34 pagesR0-Design Calculation Report of Loading FrameMuhammad Irfan ButtNo ratings yet
- TRANSIENT FLOWDocument5 pagesTRANSIENT FLOWAzizul HakimNo ratings yet
- Ejercicios Trazadores Cubicos, Metodos NumericosDocument10 pagesEjercicios Trazadores Cubicos, Metodos NumericosDaniel PlazasNo ratings yet
- Kingrynormalized Data Set AssignmentDocument24 pagesKingrynormalized Data Set Assignment20013122-001No ratings yet
- Parameter Values A12, A21, and CDocument5 pagesParameter Values A12, A21, and Cari suryo lenggonoNo ratings yet
- Eigen Values - SnapshotsDocument6 pagesEigen Values - SnapshotsVidya VNo ratings yet
- MLR-handson - Jupyter NotebookDocument5 pagesMLR-handson - Jupyter NotebookAnzal MalikNo ratings yet
- Perubahan Bobot: Perlakuan Ulangan Awal Akhir Selisih SD (%)Document9 pagesPerubahan Bobot: Perlakuan Ulangan Awal Akhir Selisih SD (%)efendiNo ratings yet
- Tensile Testing LabDocument4 pagesTensile Testing LabQ_TNo ratings yet
- Moment Influence Line ReportDocument13 pagesMoment Influence Line Reporthaziq khairudin33% (3)
- Margules Uniquac2Document12 pagesMargules Uniquac2Tias Faizatul MunirohNo ratings yet
- Taller 05 10 2020Document12 pagesTaller 05 10 2020Alejandro MalagonNo ratings yet
- Shear Force Full ReportDocument14 pagesShear Force Full ReportMoganraj93% (73)
- PCA of Covariance Matrix in MinitabDocument7 pagesPCA of Covariance Matrix in MinitabRico IndraNo ratings yet
- C B 01 Ta3 WingconfigurationDocument30 pagesC B 01 Ta3 WingconfigurationKrizelle LaoNo ratings yet
- Removing Stationarity (Beer Production Data-8years) : Vipin Himani Deepak MananDocument12 pagesRemoving Stationarity (Beer Production Data-8years) : Vipin Himani Deepak MananVM MonuNo ratings yet
- P42004 - Factor-Automobile DataDocument4 pagesP42004 - Factor-Automobile DataAbhishek PatelNo ratings yet
- Practica MRUADocument3 pagesPractica MRUAMr. Hot RodNo ratings yet
- LATIANDocument15 pagesLATIANMuhammad Maulana 24No ratings yet
- Data ScienceDocument1 pageData Science2001yashkhandelwalNo ratings yet
- Practice 24 25Document10 pagesPractice 24 25Marisnelvys CabrejaNo ratings yet
- Homework - 1 MATLAB Exercises SolutionsDocument5 pagesHomework - 1 MATLAB Exercises Solutionsmanthan212No ratings yet
- Regression ResubmitDocument7 pagesRegression Resubmitapi-3729749No ratings yet
- Examenes 201Document16 pagesExamenes 201leo rfgrNo ratings yet
- Motion Graph Data TableDocument3 pagesMotion Graph Data TableMr. Hot RodNo ratings yet
- 111-2 Data Analysis Assignment: I-cos2θ圖Document2 pages111-2 Data Analysis Assignment: I-cos2θ圖陳泓睿No ratings yet
- bobina exploradoraDocument7 pagesbobina exploradoraJUAN SEBASTIAN MOSQUERA TORRESNo ratings yet
- .5Z A ZT - A - 2.08814 - 0.36738 - 0.46584 - 1.07259 - 1.70274: - 0.08834 0.51719 - 0.33917 0.33235 0.05213Document11 pages.5Z A ZT - A - 2.08814 - 0.36738 - 0.46584 - 1.07259 - 1.70274: - 0.08834 0.51719 - 0.33917 0.33235 0.05213Haiva Qurrota A'yunNo ratings yet
- .5Z A ZT - A - 2.08814 - 0.36738 - 0.46584 - 1.07259 - 1.70274: - 0.08834 0.51719 - 0.33917 0.33235 0.05213Document11 pages.5Z A ZT - A - 2.08814 - 0.36738 - 0.46584 - 1.07259 - 1.70274: - 0.08834 0.51719 - 0.33917 0.33235 0.05213Haiva Qurrota A'yunNo ratings yet
- AutocorrelacionDocument120 pagesAutocorrelacionChristian SánchezNo ratings yet
- Getting The LogarithmDocument6 pagesGetting The Logarithmkrish lopezNo ratings yet
- Calculo Ejes Equivalentes AASHTODocument3 pagesCalculo Ejes Equivalentes AASHTOJhonn Benneth Cusacani RamosNo ratings yet
- Experimental CalculationsDocument5 pagesExperimental CalculationsmomenNo ratings yet
- Win Loss ReportDocument2 pagesWin Loss ReportRichard PaulinoNo ratings yet
- Contigencia2Document3 pagesContigencia2keveny2000No ratings yet
- Data and Model SpecificationDocument4 pagesData and Model SpecificationSakib AhmedNo ratings yet
- Output LisrelDocument26 pagesOutput LisrellustiyanidesyrNo ratings yet
- Shear and Moment Calculations for Beam SectionsDocument5 pagesShear and Moment Calculations for Beam SectionssushilNo ratings yet
- La Première ExpérienceDocument5 pagesLa Première ExpérienceMohamed El-amine BendjoudaNo ratings yet
- STRUCTURE CONTROL AND BASE ISOLATION: Numerical Solution using Newmarks MethodDocument14 pagesSTRUCTURE CONTROL AND BASE ISOLATION: Numerical Solution using Newmarks MethodRupesh UpretyNo ratings yet
- RF ImpedanceDocument7 pagesRF ImpedancesrybsantosNo ratings yet
- 202201106_lab2-2-8Document7 pages202201106_lab2-2-8duelgodgaming007No ratings yet
- PLS1Document121 pagesPLS1silviNo ratings yet
- LAB-KTKQ-Hydrodynamics of Screw PropellerDocument3 pagesLAB-KTKQ-Hydrodynamics of Screw PropellerPanda On FireNo ratings yet
- Experiment 1 - Deflected Shape: Max Max - 2 - 2Document5 pagesExperiment 1 - Deflected Shape: Max Max - 2 - 2Bilal ÇayaNo ratings yet
- Hasil Regresi Eviews Panel B - GrowthDocument7 pagesHasil Regresi Eviews Panel B - GrowthPrinika RafiuningtyasNo ratings yet
- Zub Indeterminate Truss DegDocument13 pagesZub Indeterminate Truss DegAnonymous jmo0bcjAsNo ratings yet
- IEEE 10 Generator 39 Bus System: General OutlineDocument7 pagesIEEE 10 Generator 39 Bus System: General Outlineadau100% (1)
- Equation Solving ExamplesDocument10 pagesEquation Solving Examplesalif08042012No ratings yet
- Equation Solving ExamplesDocument10 pagesEquation Solving Examplesalif08042012No ratings yet
- Newton'S Divided Difference Interpolating PolynomialsDocument3 pagesNewton'S Divided Difference Interpolating PolynomialsKatherine Shayne YeeNo ratings yet
- SIMPSONDocument367 pagesSIMPSONYatniel BjNo ratings yet
- Book 1Document60 pagesBook 1Evi diah phitalokANo ratings yet
- "Automatic" Determination of Particle Size Distribution: Analyze Set ScaleDocument6 pages"Automatic" Determination of Particle Size Distribution: Analyze Set ScaleIndalecio PrietoNo ratings yet
- AR 303 Vol 1 DDocument968 pagesAR 303 Vol 1 DAnonymous gM6RZL5lYdNo ratings yet
- n (4) * (7) ε 1 2 3 4 5 6 7 8 9 x x f (x) f (x) x = (x +x) /2 f (x)Document2 pagesn (4) * (7) ε 1 2 3 4 5 6 7 8 9 x x f (x) f (x) x = (x +x) /2 f (x)Egit Tio NandaNo ratings yet
- A. Harga Rata-Rata: N LogxiDocument17 pagesA. Harga Rata-Rata: N LogxilutviaNo ratings yet
- Dimitrova-Grajzl, Valentina Simon, Eszter. 2010. Political Trust and Historical Legacy The EffectDocument23 pagesDimitrova-Grajzl, Valentina Simon, Eszter. 2010. Political Trust and Historical Legacy The EffectsernumenserNo ratings yet
- The Ecology of Plants by Jessica GurevtichDocument465 pagesThe Ecology of Plants by Jessica GurevtichDana Ammann100% (10)
- (Poznañ Studies in the Philosophy of the Sciences and the Humanities 86) Martin R. Jones (Ed.), Nancy Cartwright (Ed.)-Idealization XII_ Correcting the Model_ Idealization and Abstraction in the ScienDocument305 pages(Poznañ Studies in the Philosophy of the Sciences and the Humanities 86) Martin R. Jones (Ed.), Nancy Cartwright (Ed.)-Idealization XII_ Correcting the Model_ Idealization and Abstraction in the Scienperception888No ratings yet
- Sampling Plan For Cleanroom ClassificationDocument15 pagesSampling Plan For Cleanroom Classificationjpabloqf100% (2)
- EcoSim 5.0 Help SystemDocument23 pagesEcoSim 5.0 Help Systemjulia0hNo ratings yet
- Chapter 1 Slides PDFDocument45 pagesChapter 1 Slides PDFjennyNo ratings yet
- Basic Statistics For Health Sciences (Third Edition) by Kuzma PDFDocument361 pagesBasic Statistics For Health Sciences (Third Edition) by Kuzma PDFFurqan Shahid67% (3)
- Analysis of Variance TableDocument19 pagesAnalysis of Variance TableJefferson AlvarezNo ratings yet
- Guided Inductive Inquiry Teaching StrategyDocument12 pagesGuided Inductive Inquiry Teaching StrategyMary Ellen Luceña100% (1)
- Machine Learning Business ReportDocument34 pagesMachine Learning Business ReportMITESH AGRAWAL100% (1)
- Family Income and Toilet Usage FactorsDocument10 pagesFamily Income and Toilet Usage FactorsShikarin KitaNo ratings yet
- MATH 121 (Chapter 10) - Correlation & RegressionDocument30 pagesMATH 121 (Chapter 10) - Correlation & RegressionpotsuNo ratings yet
- Financial Analytics Using R: Modeling Seasonality and Dummy VariablesDocument115 pagesFinancial Analytics Using R: Modeling Seasonality and Dummy Variablesshivam chugh100% (1)
- Principles of Biostatistics: Pubh605 (3 Credit Hours)Document17 pagesPrinciples of Biostatistics: Pubh605 (3 Credit Hours)eshet chafNo ratings yet
- IE005 Lab Exercise 1 and 2mnDocument7 pagesIE005 Lab Exercise 1 and 2mnApril Dawates100% (1)
- Chi Square TestDocument5 pagesChi Square TestDwarika prasad choudhuryNo ratings yet
- PENGARUH STRATEGI KELUARGA TERHADAP PASIEN HALUSINASIDocument8 pagesPENGARUH STRATEGI KELUARGA TERHADAP PASIEN HALUSINASInovanNo ratings yet
- easter-revision-day-3-workbook-new-1712407998Document40 pageseaster-revision-day-3-workbook-new-1712407998robinsonbryNo ratings yet
- Statistics: Chapter 9: Inferences Based On Two Samples: Confidence Intervals and Tests of HypothesesDocument62 pagesStatistics: Chapter 9: Inferences Based On Two Samples: Confidence Intervals and Tests of HypothesesElena WritesNo ratings yet
- Saba Anwar 1024 Analytical Chemistry.Document3 pagesSaba Anwar 1024 Analytical Chemistry.Pirate 001No ratings yet
- Statistical Data Analysis Book Dang Quang The HongDocument256 pagesStatistical Data Analysis Book Dang Quang The HongAnonymous gInYZHU100% (1)
- Assignment - Marks 60 (6X10 60) Statistics For Management Subject Code - MB0044Document12 pagesAssignment - Marks 60 (6X10 60) Statistics For Management Subject Code - MB0044Niyatitejas ParekhNo ratings yet
- T Test, ANOVA 2Document54 pagesT Test, ANOVA 2esmeralda indrianiNo ratings yet
- Unit Task - Introduction To Statistical Inference and Summary of The Different Basic Statistical Tests (I)Document3 pagesUnit Task - Introduction To Statistical Inference and Summary of The Different Basic Statistical Tests (I)Kia GraceNo ratings yet
- 2613Document41 pages2613iain-suNo ratings yet
- A Study On The Impact of Social Media On Consumer Buying Behaviour of Mobile Phones in ChennaiDocument6 pagesA Study On The Impact of Social Media On Consumer Buying Behaviour of Mobile Phones in ChennaiHeba AliNo ratings yet
- Math 1Document22 pagesMath 1RANJIT BISWAL (Ranjit)No ratings yet
- Data Analysis and Interpretation: IpdetDocument55 pagesData Analysis and Interpretation: IpdetJaved KhanNo ratings yet
- Thai Consumers Behavior Towards Fast FashionDocument6 pagesThai Consumers Behavior Towards Fast FashionMurni Ni WayanNo ratings yet
- 1960 IRE Transactions on Matched FiltersDocument19 pages1960 IRE Transactions on Matched Filtersbalaji_gawalwad9857No ratings yet