100% found this document useful (1 vote)
646 views4 pagesGuidelines for Packaging Validation
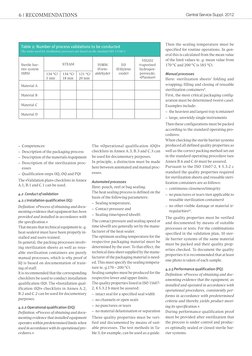
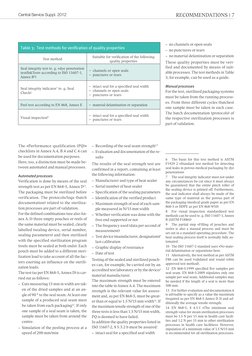
This document provides guidelines for validating packaging processes according to ISO 11607-2. It outlines the standards that provide the normative basis and prerequisites for validation. The validation process includes drafting a validation plan, qualification steps for installation, operation and performance, and a validation report. The number of validation combinations can be reduced by focusing on worst-case scenarios and choosing packaging materials appropriately. Both automated and manual packaging processes must be validated.
Uploaded by
박성민Copyright
© © All Rights Reserved
We take content rights seriously. If you suspect this is your content, claim it here.
Available Formats
Download as PDF, TXT or read online on Scribd
100% found this document useful (1 vote)
646 views4 pagesGuidelines for Packaging Validation
This document provides guidelines for validating packaging processes according to ISO 11607-2. It outlines the standards that provide the normative basis and prerequisites for validation. The validation process includes drafting a validation plan, qualification steps for installation, operation and performance, and a validation report. The number of validation combinations can be reduced by focusing on worst-case scenarios and choosing packaging materials appropriately. Both automated and manual packaging processes must be validated.
Uploaded by
박성민Copyright
© © All Rights Reserved
We take content rights seriously. If you suspect this is your content, claim it here.
Available Formats
Download as PDF, TXT or read online on Scribd