You might also like
- Working Guide to Vapor-Liquid Phase Equilibria CalculationsFrom EverandWorking Guide to Vapor-Liquid Phase Equilibria CalculationsRating: 5 out of 5 stars5/5 (1)
- Topic 14 - VaporizationDocument4 pagesTopic 14 - VaporizationiitdvivNo ratings yet
- Thermal hw1Document1 pageThermal hw1lifecrazy657No ratings yet
- CHEN 623 Problems Old Examination Problems: 1.: (P + A /V) (V B) RTDocument34 pagesCHEN 623 Problems Old Examination Problems: 1.: (P + A /V) (V B) RTZohaib Ali0% (1)
- 20 Van Der Waals Model of Liquid-Gas Phase TransitionDocument7 pages20 Van Der Waals Model of Liquid-Gas Phase TransitionAlejandro RMNo ratings yet
- Equations of StateDocument33 pagesEquations of StateDevika BharathanNo ratings yet
- Hw1phys2 2019Document3 pagesHw1phys2 2019jiddagerNo ratings yet
- Chapter14 ThermodynamicsDocument30 pagesChapter14 ThermodynamicsArvind RaveeNo ratings yet
- Lecture MMC301 Up To 05-09-2023 PDFDocument74 pagesLecture MMC301 Up To 05-09-2023 PDFashutoshranjan275No ratings yet
- Thermodynamics ShortenedDocument6 pagesThermodynamics ShortenedrasajatiNo ratings yet
- On The Dieterici Equation of StateDocument7 pagesOn The Dieterici Equation of StateRdNo ratings yet
- Lecture Notes 1 Including Fluid Properties and AnnotationsDocument43 pagesLecture Notes 1 Including Fluid Properties and AnnotationsMinjae LeeNo ratings yet
- Work in Irreversible ExpansionsDocument3 pagesWork in Irreversible ExpansionsABRAHÁM CARMELO CASTILLA SILVESTRENo ratings yet
- Chapter4-Lecture No.1Document19 pagesChapter4-Lecture No.1Mohammad SaleemNo ratings yet
- J.Chem. Ed., 1993, 70 (2), 96Document3 pagesJ.Chem. Ed., 1993, 70 (2), 96IvonneNo ratings yet
- ITK-233-2 - PVT Behavior of FluidDocument57 pagesITK-233-2 - PVT Behavior of FluidVinay GuptaNo ratings yet
- PH210x - Principles of Thermodqynamics 2Document4 pagesPH210x - Principles of Thermodqynamics 2aliyalaz100% (1)
- ThermodynamicsDocument32 pagesThermodynamicsAsim AnsariNo ratings yet
- Mathematical Modeling of Chemical ProcessesDocument12 pagesMathematical Modeling of Chemical ProcessesSeanne CruzNo ratings yet
- ThermodynamicsDocument14 pagesThermodynamicsPurpleNo ratings yet
- Hermodynamics of Phase Change: Hase ChangesDocument14 pagesHermodynamics of Phase Change: Hase ChangesdemirciNo ratings yet
- Ps 1Document2 pagesPs 1Ram Balaji100% (1)
- Lecture 3a - Non-Ideal Thermal Equations of StateDocument6 pagesLecture 3a - Non-Ideal Thermal Equations of StateMuhammad Usman Saifullah KhanNo ratings yet
- Origin of The A/v Term: 2 M Moles MolesDocument5 pagesOrigin of The A/v Term: 2 M Moles MolesNandNNo ratings yet
- Phase Transitions - Lecture NotesDocument51 pagesPhase Transitions - Lecture NotesKhaled TamimyNo ratings yet
- Lecture5 - Final Stat MechDocument21 pagesLecture5 - Final Stat MechnokosamNo ratings yet
- We Shall Make The Following AssumptionsDocument6 pagesWe Shall Make The Following AssumptionsMuhammad Fatah KaryadiNo ratings yet
- Lecture Note 12Document53 pagesLecture Note 12Mitsuha IzuyamiNo ratings yet
- The Van Der Waals' Gas (VDW.G.) : NRT RT P V VDocument5 pagesThe Van Der Waals' Gas (VDW.G.) : NRT RT P V VSoji AdimulaNo ratings yet
- Typical Examples of Irreversible ProcessesDocument8 pagesTypical Examples of Irreversible ProcessesadminchemNo ratings yet
- Class 11 Physics Revision Notes ThermodynamicsDocument35 pagesClass 11 Physics Revision Notes ThermodynamicsSwastika DasNo ratings yet
- Improvement of The Van Der Waals Equation of StateDocument13 pagesImprovement of The Van Der Waals Equation of StateRené Mora-CasalNo ratings yet
- PV Work TermDocument4 pagesPV Work TermvaraduNo ratings yet
- A Comparison of Equations of StateDocument8 pagesA Comparison of Equations of StateDarren Sean HoNo ratings yet
- Statistical Physics by VeselyDocument71 pagesStatistical Physics by VeselycrguntalilibNo ratings yet
- Saturation Pressure FoysDocument6 pagesSaturation Pressure FoysKalson UmpuNo ratings yet
- Chemical Engineering ThermodynamicsDocument5 pagesChemical Engineering ThermodynamicsP P DNo ratings yet
- Latent Heat of Vaporization of EthanolDocument5 pagesLatent Heat of Vaporization of EthanolMel DyNo ratings yet
- Virial Equation of StateDocument9 pagesVirial Equation of StateSaba ArifNo ratings yet
- KTG Part 1Document9 pagesKTG Part 1Sarvan SankaranNo ratings yet
- Ch4 Closed SystemDocument10 pagesCh4 Closed SystemEpimerianos AberianosNo ratings yet
- Tut 04Document2 pagesTut 04Ebert AroneNo ratings yet
- The Van Der Waals Equation Analytical and Approximate SolutionsDocument21 pagesThe Van Der Waals Equation Analytical and Approximate Solutionsvitoribeiro90No ratings yet
- Chap 3 - Volumetric Properties of FluidDocument38 pagesChap 3 - Volumetric Properties of FluidHonestNo ratings yet
- Chem 14 Laboratory Report Van Der Waals Isotherms 5 PDF FreeDocument6 pagesChem 14 Laboratory Report Van Der Waals Isotherms 5 PDF FreePedro Ian QuintanillaNo ratings yet
- CHCC 117 Adiabatic NotesDocument3 pagesCHCC 117 Adiabatic NotesSkye DiazNo ratings yet
- General Heat Transport Equation Heat Transfer CoefficientDocument6 pagesGeneral Heat Transport Equation Heat Transfer CoefficientasdfghjkhNo ratings yet
- AssignmentDocument2 pagesAssignmentshamik dattaNo ratings yet
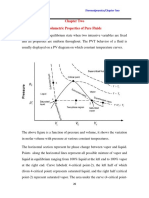
- Chapter Two Volumetric Properties of Pure FluidsDocument18 pagesChapter Two Volumetric Properties of Pure Fluidsc74zdwfbzhNo ratings yet
- Transient Flow Analysis:: DR - Nasiruddinshaikh Advanced Thermodynamics (Me 541)Document7 pagesTransient Flow Analysis:: DR - Nasiruddinshaikh Advanced Thermodynamics (Me 541)Nadeem TanwariNo ratings yet
- Thermodynamics 1: Volumetric Properties of Pure FluidsDocument24 pagesThermodynamics 1: Volumetric Properties of Pure FluidsHabib Faisal Yahya100% (1)
- Chapter Two Volumetric Properties of Pure FluidsDocument18 pagesChapter Two Volumetric Properties of Pure Fluidsson gokuNo ratings yet
- CHE325 Note1 From DR AyoolaDocument30 pagesCHE325 Note1 From DR AyoolaPreciousNo ratings yet
- Gas Dynamics: Instructor: Dr. AsadullahDocument12 pagesGas Dynamics: Instructor: Dr. AsadullahM Rakeeb BalochNo ratings yet
- Enstrophy Dissipation in Two-Dimensional TurbulenceDocument7 pagesEnstrophy Dissipation in Two-Dimensional TurbulenceLuis CardenasNo ratings yet
- Lecture 2Document21 pagesLecture 2Ahmed SajjadNo ratings yet
- Lecture 02Document9 pagesLecture 02Putu IndraNo ratings yet
- Claysius Clapeyron Lab ExperimentDocument11 pagesClaysius Clapeyron Lab Experimentmohamad munzir100% (1)
- Introduction and Properties of Pure SubstancesDocument63 pagesIntroduction and Properties of Pure SubstancesTushyNo ratings yet
- Chapter 3Document13 pagesChapter 3Kev AdiNo ratings yet
- LP Science2 q2w5Document7 pagesLP Science2 q2w5Gaila Mae Abejuela SanorjoNo ratings yet
- Famous Landmarks Vocabulary Cards Classroom Posters Conversation Topics Dialogs Flas - 72196Document1 pageFamous Landmarks Vocabulary Cards Classroom Posters Conversation Topics Dialogs Flas - 72196lolita digiacomo100% (1)
- Antimicrobial Spectrum and Characteristics of Hand-Hygiene Antiseptic AgentsDocument3 pagesAntimicrobial Spectrum and Characteristics of Hand-Hygiene Antiseptic AgentsdonsterthemonsterNo ratings yet
- FrecklesDocument12 pagesFrecklesEnoque PinheiroNo ratings yet
- Scope of Work For FEED - Table of ContentsDocument5 pagesScope of Work For FEED - Table of ContentsFaizal Sattu100% (7)
- RoyalBritishBank V TurquanDocument3 pagesRoyalBritishBank V TurquanAlbukhari AliasNo ratings yet
- Midview of 2Document4 pagesMidview of 2api-285760777No ratings yet
- Bài Kt 2 Biên Dịch 1-LiêmDocument10 pagesBài Kt 2 Biên Dịch 1-LiêmNguyen Loan100% (1)
- Corporation: Pre-Trimmed IC Voltage Controlled AmplifiersDocument8 pagesCorporation: Pre-Trimmed IC Voltage Controlled AmplifiersBrandon ParsonsNo ratings yet
- Assignment Arts Appreciation: Submitted To: Mam Sana Submitted By: Mahnoor Akhter Session: 2018-2022 Semester: 1Document16 pagesAssignment Arts Appreciation: Submitted To: Mam Sana Submitted By: Mahnoor Akhter Session: 2018-2022 Semester: 1Mahnoor AkhterNo ratings yet
- Chapter 18 Foundation of ControlDocument25 pagesChapter 18 Foundation of Controlhalka2314No ratings yet
- Good Dissertation Topics For Sports TherapyDocument5 pagesGood Dissertation Topics For Sports TherapyPapersHelpEvansville100% (1)
- E-Katalog Warna - FullDocument43 pagesE-Katalog Warna - Fullarend alfiyantoNo ratings yet
- Case Study MtotDocument15 pagesCase Study MtotDotecho Jzo EyNo ratings yet
- Master Teacher Developmental Plan Real2021 2022Document6 pagesMaster Teacher Developmental Plan Real2021 2022Roygvib Clemente MontañoNo ratings yet
- Vietnam Maritime UniversityDocument36 pagesVietnam Maritime UniversitySơn BáchNo ratings yet
- On Three Monotone Approximation: Mayada Ali KareemDocument4 pagesOn Three Monotone Approximation: Mayada Ali KareemMohamed Aly SowNo ratings yet
- Case Study Analysis On Reinventing The Wheel at Apex Door CompanyDocument5 pagesCase Study Analysis On Reinventing The Wheel at Apex Door CompanySivachandran RNo ratings yet
- Decision Tree PrimerDocument42 pagesDecision Tree PrimerMuzzamil JanjuaNo ratings yet
- International Journal of Transportation Science and TechnologyDocument11 pagesInternational Journal of Transportation Science and TechnologyIrvin SmithNo ratings yet
- Your Ights: MR Simrat Singh ARORADocument4 pagesYour Ights: MR Simrat Singh ARORAsimratsingh01No ratings yet
- Penilaian Kelayakan Usaha Mikro Dengan Kredit Skoring Dan Pengaruhnya Terhadap Pembiayaan Bermasalah Best Practice Lembaga Keuangan Di IndonesiaDocument12 pagesPenilaian Kelayakan Usaha Mikro Dengan Kredit Skoring Dan Pengaruhnya Terhadap Pembiayaan Bermasalah Best Practice Lembaga Keuangan Di IndonesiaRiantriaNo ratings yet
- Unit 8 Standard Test Test (Z Widoczną Punktacją)Document6 pagesUnit 8 Standard Test Test (Z Widoczną Punktacją)lucjakrzywa2804No ratings yet
- Bipolar MembraneDocument3 pagesBipolar MembranepriyaNo ratings yet
- Road EstimateDocument15 pagesRoad EstimateRAJ KNo ratings yet
- Volkswagen 2010 Maintenance ScheduleDocument4 pagesVolkswagen 2010 Maintenance ScheduleWillNo ratings yet
- GRADE 9-National Reading ProgramDocument2 pagesGRADE 9-National Reading ProgramJulius Bayaga100% (1)
- 40 Questions in 23 Minutes - Maths by Arun SirDocument42 pages40 Questions in 23 Minutes - Maths by Arun SirSatya RanjanNo ratings yet
- CFA Level I - Timetable (August 2021 Exam) (V2)Document1 pageCFA Level I - Timetable (August 2021 Exam) (V2)Via Commerce Sdn BhdNo ratings yet
- REC4281GDocument307 pagesREC4281GadrianahoukiNo ratings yet