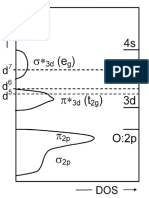
You might also like
- Advanced Functional Materials: A Perspective from Theory and ExperimentFrom EverandAdvanced Functional Materials: A Perspective from Theory and ExperimentNo ratings yet
- Acs Inorgchem 7b02272Document12 pagesAcs Inorgchem 7b02272SETIFI FatimaNo ratings yet
- Reviews in Computational ChemistryFrom EverandReviews in Computational ChemistryAbby L. ParrillNo ratings yet
- Plasmonic Physics of 2D Crystalline Materials ExplainedDocument30 pagesPlasmonic Physics of 2D Crystalline Materials Explained유재원No ratings yet
- Spin-Flip Reactions of ZR + C H Researched by Relativistic Density Functional TheoryDocument10 pagesSpin-Flip Reactions of ZR + C H Researched by Relativistic Density Functional TheoryDiego Alejandro Hurtado BalcazarNo ratings yet
- High Temperature and Pressure DependentDocument17 pagesHigh Temperature and Pressure DependentAlam KhanNo ratings yet
- PhysRevB 101 224430Document6 pagesPhysRevB 101 224430Kamini GautamNo ratings yet
- Science Abc6363Document5 pagesScience Abc6363NeerajNo ratings yet
- Quasiparticle Dynamics in GrapheneDocument6 pagesQuasiparticle Dynamics in GrapheneThamyres F. Messa MoreiraNo ratings yet
- Spectroscopic and Theoretical AspectsDocument123 pagesSpectroscopic and Theoretical AspectsMPCNo ratings yet
- Band-Selective Holstein Polaron in Luttinger Liquid Material A Moo (A K, RB)Document7 pagesBand-Selective Holstein Polaron in Luttinger Liquid Material A Moo (A K, RB)Scribd ScribdNo ratings yet
- Crystals 11 01050 v2Document7 pagesCrystals 11 01050 v2Giovanni Samir Perez HerreraNo ratings yet
- Physica E: Low-Dimensional Systems and Nanostructures: SciencedirectDocument7 pagesPhysica E: Low-Dimensional Systems and Nanostructures: SciencedirectFadjar MulyaNo ratings yet
- Density Functional Theory and Experimental Study of The Electronic Structure and Transport Properties of La, V, NB, and Ta Doped Srtio3Document12 pagesDensity Functional Theory and Experimental Study of The Electronic Structure and Transport Properties of La, V, NB, and Ta Doped Srtio3Shridhar MathadNo ratings yet
- Emission and recombination coefficients for hydrogen with κ-distributed electron energiesDocument3 pagesEmission and recombination coefficients for hydrogen with κ-distributed electron energiesjameswhite4321No ratings yet
- Ab Initio Study of The Emissive Charge-Transfer States of Solvated Chromophore-Functionalized SilsesquioxanesDocument4 pagesAb Initio Study of The Emissive Charge-Transfer States of Solvated Chromophore-Functionalized Silsesquioxanes蔡承德No ratings yet
- I. Turek Et Al - Exchange Interactions, Spin Waves, and Transition Temperatures in Itinerant MagnetsDocument36 pagesI. Turek Et Al - Exchange Interactions, Spin Waves, and Transition Temperatures in Itinerant MagnetsTellusz4532No ratings yet
- Ab Initio and Long-Range Studies of The Electronic Transition Dipole Moments Among The Low-Lying States of Rb2 and Cs2 MoleculesDocument8 pagesAb Initio and Long-Range Studies of The Electronic Transition Dipole Moments Among The Low-Lying States of Rb2 and Cs2 MoleculesCattNo ratings yet
- Paper 2Document8 pagesPaper 2Alexander AndradeNo ratings yet
- How High The Spin Allowed and Forbidden Spin StatesDocument9 pagesHow High The Spin Allowed and Forbidden Spin StatesThiago Costa SerraNo ratings yet
- Oxygen Transport in Perovskite-Type Solid Oxide Fuel Cell Materials: Insights From Quantum MechanicsDocument9 pagesOxygen Transport in Perovskite-Type Solid Oxide Fuel Cell Materials: Insights From Quantum MechanicsrajanadarajanNo ratings yet
- 4 PhysRevB.103.134502 PDFDocument7 pages4 PhysRevB.103.134502 PDF황주영No ratings yet
- PhysRevLett 117 215502Document7 pagesPhysRevLett 117 215502JOSE CARLOS LEON GONZALEZNo ratings yet
- (NDWP1D) Schlagheck Epjd03Document15 pages(NDWP1D) Schlagheck Epjd03John LeónNo ratings yet
- Ncomms 12715Document9 pagesNcomms 12715黃奕軒No ratings yet
- T. Masuda Et Al - Cooperative Ordering of Gapped and Gapless Spin Networks in Cu2Fe2Ge4O13Document4 pagesT. Masuda Et Al - Cooperative Ordering of Gapped and Gapless Spin Networks in Cu2Fe2Ge4O13Tellusz4532No ratings yet
- Stavros C. Farantos, Shi Ying Lin and Hua Guo - A Regular Isomerization Path Among Chaotic Vibrational States of CH2 (A A1)Document6 pagesStavros C. Farantos, Shi Ying Lin and Hua Guo - A Regular Isomerization Path Among Chaotic Vibrational States of CH2 (A A1)ImasmzNo ratings yet
- Sluggish Diffusion in Co-Cr-Fe-Mn-Ni High-Entropy Alloys: K.-Y. Tsai, M.-H. Tsai, J.-W. YehDocument11 pagesSluggish Diffusion in Co-Cr-Fe-Mn-Ni High-Entropy Alloys: K.-Y. Tsai, M.-H. Tsai, J.-W. YehnonameNo ratings yet
- Kinetic Prediction of Reverse Intersystem Crossing in Organic Donor - Acceptor MoleculesDocument6 pagesKinetic Prediction of Reverse Intersystem Crossing in Organic Donor - Acceptor Moleculesd20vipsplayNo ratings yet
- Tuning Magnetic Properties of Diamagnetic MoleculesDocument35 pagesTuning Magnetic Properties of Diamagnetic MoleculesRishu KhuranaNo ratings yet
- Studies On The Gamma Radiation Responses of High TC SuperconductorsDocument27 pagesStudies On The Gamma Radiation Responses of High TC Superconductorsdeni.sttnNo ratings yet
- Storey Sochi 2012Document13 pagesStorey Sochi 2012taha_sochiNo ratings yet
- 1405.6290Document32 pages1405.6290ShrabaniPaulNo ratings yet
- Density Functional Theory and Solvation ModelDocument37 pagesDensity Functional Theory and Solvation Modelsiska tasyaNo ratings yet
- PhysRevB.100.235420 - PbTaSe2Document8 pagesPhysRevB.100.235420 - PbTaSe2Jeon LeoiNo ratings yet
- Effect of Disorder on Heavy Electron Fermi SurfacesDocument6 pagesEffect of Disorder on Heavy Electron Fermi SurfacesYoshio KuramotoNo ratings yet
- 2016 Chem. Phys. Vibrational Effects of Charging OPD1Document8 pages2016 Chem. Phys. Vibrational Effects of Charging OPD1Chris SmithNo ratings yet
- M. Takahashi Et Al - Effective Field Theory For Spinor Dipolar Bose Einstein CondensatesDocument11 pagesM. Takahashi Et Al - Effective Field Theory For Spinor Dipolar Bose Einstein CondensatesPomac232No ratings yet
- Book WikiDocument68 pagesBook Wikialice.medeirosNo ratings yet
- Par-16-Mapping Atomic OrbitalsDocument5 pagesPar-16-Mapping Atomic OrbitalssarizbethcortesgarciaNo ratings yet
- Molecular Transport Junctions: Vibrational EffectsDocument118 pagesMolecular Transport Junctions: Vibrational EffectsAbdalmoedAlaiashyNo ratings yet
- Article-Numerical Study of Reactive Radical Species Formation in Methane Plasma DischargeDocument6 pagesArticle-Numerical Study of Reactive Radical Species Formation in Methane Plasma DischargeKhawlaNo ratings yet
- Phsv01i01p0069 PDFDocument6 pagesPhsv01i01p0069 PDFphysicsjournalNo ratings yet
- Crystals 11 01109Document8 pagesCrystals 11 01109hamza bakkaliNo ratings yet
- Reactivity of Indoles Through The Eyes of A Charge-Transfer Partitioning AnalysisDocument7 pagesReactivity of Indoles Through The Eyes of A Charge-Transfer Partitioning AnalysistrabajosNo ratings yet
- Stacking Fault Energies of Face-Centered Cubic Concentrated Solid Solution AlloysDocument29 pagesStacking Fault Energies of Face-Centered Cubic Concentrated Solid Solution AlloysNuzulul RahmahNo ratings yet
- Negative Differential Resistance in NanoDocument9 pagesNegative Differential Resistance in NanoTobiasNo ratings yet
- Gin Dens Per Ger 2010Document7 pagesGin Dens Per Ger 2010Diana UrizaNo ratings yet
- PhysRevD 99 052002 PDFDocument28 pagesPhysRevD 99 052002 PDFBibhuprasad MahakudNo ratings yet
- HH 2020Document13 pagesHH 2020Djallal Eddine MellahNo ratings yet
- Lab 4 - MODULE γ-2: 3.014 Materials Laboratory Dec. 8 - Dec..13, 2006Document8 pagesLab 4 - MODULE γ-2: 3.014 Materials Laboratory Dec. 8 - Dec..13, 2006ALEXANDER DAVID PARICELA CRUZNo ratings yet
- C-PCM Study of Solvent Polarity Effect On Spin Crossover in Complex (NCS) )Document7 pagesC-PCM Study of Solvent Polarity Effect On Spin Crossover in Complex (NCS) )ALBA NIETONo ratings yet
- Local Criticality, Diffusion and Chaos in Generalized Sachdev-Ye-Kitaev ModelsDocument38 pagesLocal Criticality, Diffusion and Chaos in Generalized Sachdev-Ye-Kitaev ModelspiNo ratings yet
- Spin-Forbidden Transitions in The Spectra of Transition Metal Ions and Nephelauxetic EffectDocument11 pagesSpin-Forbidden Transitions in The Spectra of Transition Metal Ions and Nephelauxetic EffectDaniela Araújo RodríguezNo ratings yet
- 1 s2.0 S0042207X23008746 MainDocument6 pages1 s2.0 S0042207X23008746 MainAamir ShafiqueNo ratings yet
- Solid State Calculations Using WIEN2kDocument15 pagesSolid State Calculations Using WIEN2kabu_dzulfiqar5528100% (1)
- Two Particle Spin Dependent Interactions in Hgte/Cdte Topological InsulatorsDocument9 pagesTwo Particle Spin Dependent Interactions in Hgte/Cdte Topological InsulatorsMarius A RaduNo ratings yet
- Electronic G Tensors in U Complexes-A Computational Study: DOI: 10.1002/chem.201701058Document11 pagesElectronic G Tensors in U Complexes-A Computational Study: DOI: 10.1002/chem.201701058Austin LloydNo ratings yet
- Theoretical study of TPA properties of octupolar metal complexesDocument10 pagesTheoretical study of TPA properties of octupolar metal complexesyalocim666No ratings yet
- Monoelectronic Band Structure For A Cubic PerovskiteDocument1 pageMonoelectronic Band Structure For A Cubic PerovskiteSJNo ratings yet
- CHM510 - SpeDocument7 pagesCHM510 - SpeafifiNo ratings yet
- BS en Iso 945 Part 3Document52 pagesBS en Iso 945 Part 3vkkt2016No ratings yet
- Casio AT 1 Service ManualDocument28 pagesCasio AT 1 Service ManualMario Gabriel MoralliNo ratings yet
- EngineeringDocument8 pagesEngineeringkumarNo ratings yet
- Molflow User GuideDocument79 pagesMolflow User Guidemarian56No ratings yet
- Saptarshi PDFDocument15 pagesSaptarshi PDFViriato SouzaNo ratings yet
- D5G6 DatasheetDocument3 pagesD5G6 DatasheetchichedemorenoNo ratings yet
- An Introduction To Probability Theory and Its Applications (Vol.1), Feller WDocument525 pagesAn Introduction To Probability Theory and Its Applications (Vol.1), Feller WKayalvizhi SelvamNo ratings yet
- Chapter 3 Kinematics in Two Dimensions VectorsDocument18 pagesChapter 3 Kinematics in Two Dimensions VectorsShaquille AbhistaNo ratings yet
- WIMO Final 2018 Mock P1fDocument8 pagesWIMO Final 2018 Mock P1fDo Yun100% (1)
- NadIR v1.0.0 User Manual PDFDocument10 pagesNadIR v1.0.0 User Manual PDFPaul GarridoNo ratings yet
- Deep Learning For Sentiment Analysis of Tunisian DDocument21 pagesDeep Learning For Sentiment Analysis of Tunisian DJamila HamdiNo ratings yet
- Cashflow STS User Manual - G2Document46 pagesCashflow STS User Manual - G2AleksandarNo ratings yet
- AMETANK REPORT: Roof design calculationsDocument41 pagesAMETANK REPORT: Roof design calculationsHasan arif KısaalioğluNo ratings yet
- MK300 - Catalogue Relay bảo vệ dòng rò (Earth Leakage Relay) MK300 của MikroDocument2 pagesMK300 - Catalogue Relay bảo vệ dòng rò (Earth Leakage Relay) MK300 của MikroconmavtvnNo ratings yet
- LAS EIM 9-Week 2Document12 pagesLAS EIM 9-Week 2jon pantzNo ratings yet
- Manual Imo Pump 3g SeriesDocument8 pagesManual Imo Pump 3g SeriesВладимир50% (2)
- Management ScienceDocument3 pagesManagement ScienceRevenlie GalapinNo ratings yet
- Chemistry Paper (12 TH New Pattern)Document2 pagesChemistry Paper (12 TH New Pattern)Suyog TekamNo ratings yet
- NEF Tier 3 Electronic EngineDocument120 pagesNEF Tier 3 Electronic EngineJuan Guzmán100% (12)
- Delhi Public School, Ranchi: Coordination CompoundsDocument1 pageDelhi Public School, Ranchi: Coordination CompoundsScience SpiritNo ratings yet
- Calculation and Spesification of Fuel Oil System: Design Iv Machinery Department of Marine EngineeringDocument42 pagesCalculation and Spesification of Fuel Oil System: Design Iv Machinery Department of Marine EngineeringMuzami ThahirNo ratings yet
- Analysis and Modification of Bogie Suspension SystemDocument76 pagesAnalysis and Modification of Bogie Suspension SystemKrishnaSingh0% (2)
- Advanced DyeDocument3 pagesAdvanced DyeWalter NgoNo ratings yet
- Chemistry Webquest Introduction to AtomsDocument3 pagesChemistry Webquest Introduction to AtomsMarx GomesNo ratings yet
- DENSO A/C ComponentsDocument478 pagesDENSO A/C ComponentsPapp Ilona100% (1)
- PV776-20 003739Document0 pagesPV776-20 003739Registr Registr100% (1)
- D275A-2 Up Shop ManualDocument652 pagesD275A-2 Up Shop ManualHugo Valdes Barrios100% (11)
- Wireless - Zone Na Alarmna Centrala Dodatok Za Model 1565Document3 pagesWireless - Zone Na Alarmna Centrala Dodatok Za Model 1565billwillis66No ratings yet
- AntennasDocument5 pagesAntennasMiguel Ferrando Rocher100% (1)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (3)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Guidelines for Asset Integrity ManagementFrom EverandGuidelines for Asset Integrity ManagementRating: 5 out of 5 stars5/5 (1)
- Meltdown: Nuclear disaster and the human cost of going criticalFrom EverandMeltdown: Nuclear disaster and the human cost of going criticalRating: 5 out of 5 stars5/5 (5)
- The Periodic Table of Elements - Post-Transition Metals, Metalloids and Nonmetals | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Post-Transition Metals, Metalloids and Nonmetals | Children's Chemistry BookNo ratings yet
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 5 out of 5 stars5/5 (1)
- Chemistry at Home - A Collection of Experiments and Formulas for the Chemistry EnthusiastFrom EverandChemistry at Home - A Collection of Experiments and Formulas for the Chemistry EnthusiastNo ratings yet
- Essential Oil Chemistry Formulating Essential Oil Blends that Heal - Aldehyde - Ketone - Lactone: Healing with Essential OilFrom EverandEssential Oil Chemistry Formulating Essential Oil Blends that Heal - Aldehyde - Ketone - Lactone: Healing with Essential OilRating: 5 out of 5 stars5/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- An Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksFrom EverandAn Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksRating: 5 out of 5 stars5/5 (1)
- Coating and Drying Defects: Troubleshooting Operating ProblemsFrom EverandCoating and Drying Defects: Troubleshooting Operating ProblemsRating: 5 out of 5 stars5/5 (1)
- Science Goes Viral: Captivating Accounts of Science in Everyday LifeFrom EverandScience Goes Viral: Captivating Accounts of Science in Everyday LifeRating: 5 out of 5 stars5/5 (1)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableFrom EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableRating: 3.5 out of 5 stars3.5/5 (22)
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)
- Gas-Liquid And Liquid-Liquid SeparatorsFrom EverandGas-Liquid And Liquid-Liquid SeparatorsRating: 3.5 out of 5 stars3.5/5 (3)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeRating: 4 out of 5 stars4/5 (9)
- Stuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldFrom EverandStuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldRating: 4 out of 5 stars4/5 (289)
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (3)
- Chemical Elements Pocket Guide: Detailed Summary of the Periodic TableFrom EverandChemical Elements Pocket Guide: Detailed Summary of the Periodic TableNo ratings yet