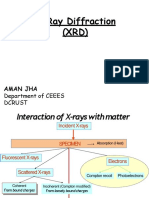
You might also like
- Chapter-13 Assessment Gases Student EditableDocument5 pagesChapter-13 Assessment Gases Student EditableFabricio FernandezNo ratings yet
- Fluid Statics Problem Set 3aDocument11 pagesFluid Statics Problem Set 3aRyan Shane Langit50% (2)
- 5 STR DeterminationTech 5oct2020Document22 pages5 STR DeterminationTech 5oct2020things strangerNo ratings yet
- X Ray CrystallographyDocument28 pagesX Ray Crystallographylexus05No ratings yet
- X-ray diffraction 7Document11 pagesX-ray diffraction 7alina.tlekkabylova270202No ratings yet
- Catalyst Characterization TechniquesDocument8 pagesCatalyst Characterization TechniquesDaniel DadzieNo ratings yet
- x Ray ScatteringDocument33 pagesx Ray ScatteringSunita PradhanNo ratings yet
- Theory: Crystallography Is The Experimental Science of Determining The Arrangement of Atoms in CrystallineDocument4 pagesTheory: Crystallography Is The Experimental Science of Determining The Arrangement of Atoms in CrystallineKyla Claire BiñasNo ratings yet
- Report XRD 8153Document16 pagesReport XRD 8153Rishabh SoodNo ratings yet
- Single Crystal X Ray DiffractionDocument4 pagesSingle Crystal X Ray Diffractioncutecat985351No ratings yet
- X-ray Crystallography ExplainedDocument55 pagesX-ray Crystallography Explainedxanadu wannaduNo ratings yet
- X-ray Crystallography: Protein Structure Determination Using X-Ray DiffractionDocument30 pagesX-ray Crystallography: Protein Structure Determination Using X-Ray Diffractionariba khanNo ratings yet
- Single-crystal X-ray Diffraction TechniqueDocument3 pagesSingle-crystal X-ray Diffraction TechniqueJosé Francisco Blanco VillalbaNo ratings yet
- MS&E 11 Raja Muhammad Usama Muhammad Asim Harris Hassan Usman Ahmed SiddiquiDocument28 pagesMS&E 11 Raja Muhammad Usama Muhammad Asim Harris Hassan Usman Ahmed SiddiquiShahZaib AnwarNo ratings yet
- Unit 4 Structure Unfolding TechniquesDocument29 pagesUnit 4 Structure Unfolding TechniquesvijayNo ratings yet
- What is Single-crystal X-ray DiffractionDocument5 pagesWhat is Single-crystal X-ray DiffractionYousef Adel HassanenNo ratings yet
- Crystal, Powder and ApplicationsDocument14 pagesCrystal, Powder and ApplicationssowjanyaNo ratings yet
- Material S. Presentation 1Document77 pagesMaterial S. Presentation 1Dagmawi HailuNo ratings yet
- Single-crystal X-ray Diffraction TechniqueDocument6 pagesSingle-crystal X-ray Diffraction TechniqueDolih Gozali100% (1)
- Electron Diffraction Using Selected Area Diffraction Pattern (SADP) in TEMDocument6 pagesElectron Diffraction Using Selected Area Diffraction Pattern (SADP) in TEMAtul DwivediNo ratings yet
- XRD (X - Ray Diffraction)Document7 pagesXRD (X - Ray Diffraction)summi64No ratings yet
- X-Ray DiffractionDocument13 pagesX-Ray Diffractionargie joy marieNo ratings yet
- Fundamentals X-Ray DiffractionDocument14 pagesFundamentals X-Ray DiffractionArif MamonNo ratings yet
- XRD Analysis for Crystal Structure DeterminationDocument15 pagesXRD Analysis for Crystal Structure DeterminationAman JhaNo ratings yet
- Comparison of Crystallography, NMR, cryoEMDocument9 pagesComparison of Crystallography, NMR, cryoEMHằngHamHốNo ratings yet
- X Ray Crystallography and Its ApplicatioDocument13 pagesX Ray Crystallography and Its ApplicatioManoj PrakashNo ratings yet
- MS&E 11 Raja Muhammad Usama Muhammad Asim Harris Hassan Usman Ahmed SiddiquiDocument28 pagesMS&E 11 Raja Muhammad Usama Muhammad Asim Harris Hassan Usman Ahmed SiddiquiShahZaib AnwarNo ratings yet
- Investigation of Fine Structure of Textile FibreDocument12 pagesInvestigation of Fine Structure of Textile FibreSuyash Manmohan100% (1)
- The Physical Structure of FiberDocument7 pagesThe Physical Structure of FiberTowfic Aziz KanonNo ratings yet
- Xray DiffractionDocument3 pagesXray DiffractionOsuizugbe Esom kennethNo ratings yet
- X-Ray Crystallography and Its Applications in Dairy Science: A ReviewDocument13 pagesX-Ray Crystallography and Its Applications in Dairy Science: A Reviewkamal gandhiNo ratings yet
- X-Ray Crystallography X-RayDocument10 pagesX-Ray Crystallography X-RayJoriel SolenonNo ratings yet
- Protein Crystallography A Concise Guide - Lattman & LollDocument149 pagesProtein Crystallography A Concise Guide - Lattman & LollAndresNo ratings yet
- LatticeDocument15 pagesLatticemisganawsimachew5No ratings yet
- XRD ReportDocument13 pagesXRD ReportMukulNo ratings yet
- X Ray DiffractionDocument12 pagesX Ray DiffractionSiddraKhalidNo ratings yet
- Material Report 1 StructuralallallDocument11 pagesMaterial Report 1 Structuralallallyassin elwakilNo ratings yet
- Sir William Henry Bragg: Noble Prize 1915!Document53 pagesSir William Henry Bragg: Noble Prize 1915!Harneysia JaneNo ratings yet
- Dr. Ashutosh Pandey Chemistry Department MNNIT AllahabadDocument22 pagesDr. Ashutosh Pandey Chemistry Department MNNIT AllahabadbotiwaNo ratings yet
- X-Ray CrystallographyDocument10 pagesX-Ray CrystallographyMuskan WahabNo ratings yet
- Neutron Diffraction and Comparison With X-Ray and Electron DiffractionDocument35 pagesNeutron Diffraction and Comparison With X-Ray and Electron DiffractionShalini BaruahNo ratings yet
- Characterization of NanoparticlesDocument16 pagesCharacterization of NanoparticlesPrasun SarkarNo ratings yet
- XRD Analysis of CNT/PAni CompositeDocument4 pagesXRD Analysis of CNT/PAni CompositeTush RohNo ratings yet
- Characterization of Nanoparticles: DR - Rabia RazzaqDocument40 pagesCharacterization of Nanoparticles: DR - Rabia RazzaqHafsa MansoorNo ratings yet
- Rigaku Journal 32-2-35-43Document9 pagesRigaku Journal 32-2-35-43Acácio CruzNo ratings yet
- XRD Analysis of Diamond Crystal StructureDocument32 pagesXRD Analysis of Diamond Crystal StructureamnaNo ratings yet
- Preparation of Dichlorobis - (Ethylenediamine) Cobalt (Iii) Chloride and Characterization With Single Crystal X-Ray DiffractionDocument7 pagesPreparation of Dichlorobis - (Ethylenediamine) Cobalt (Iii) Chloride and Characterization With Single Crystal X-Ray DiffractionJ Mora GañanNo ratings yet
- Adobe Third Party NoticesDocument15 pagesAdobe Third Party NoticesJocelyn CyrNo ratings yet
- XRDDocument27 pagesXRDBaraliya Jagdish DNo ratings yet
- Xray CrystallographyDocument33 pagesXray CrystallographyrotisatoseNo ratings yet
- DRAFT Physical Methods in Biology and Medicine Biophysics Finals RequirementsDocument16 pagesDRAFT Physical Methods in Biology and Medicine Biophysics Finals RequirementsALNo ratings yet
- Lecture TEMDocument20 pagesLecture TEMKevin McNallyNo ratings yet
- Metal Detection by XRDDocument29 pagesMetal Detection by XRDDeb RathNo ratings yet
- 04 X-Ray DiffractionDocument9 pages04 X-Ray DiffractionSampath KumarNo ratings yet
- Physical Techniques in Inorganic Chemistry: Diffraction MethodsDocument5 pagesPhysical Techniques in Inorganic Chemistry: Diffraction MethodsjorgeeduardogarciaNo ratings yet
- RFDC SputteringDocument4 pagesRFDC Sputteringसुरेन्द्र वशिष्ठNo ratings yet
- Che 323 Solved Problem Set Solid State Chemistry: Group Member Contribution Member's SignatureDocument12 pagesChe 323 Solved Problem Set Solid State Chemistry: Group Member Contribution Member's SignatureZhu Chen ChuanNo ratings yet
- III Growth of L-Proline Sodium Sulphate Single Crystals:) Group in Them (27) .Especially Some Amino Acid LikeDocument16 pagesIII Growth of L-Proline Sodium Sulphate Single Crystals:) Group in Them (27) .Especially Some Amino Acid LikeramNo ratings yet
- X-Ray Crystallography Is A Method of Determining The Arrangement ofDocument18 pagesX-Ray Crystallography Is A Method of Determining The Arrangement ofMithesh PhadtareNo ratings yet
- Introduction To Single Crystal X-Ray AnalysisDocument5 pagesIntroduction To Single Crystal X-Ray AnalysisAlejandro Altamirano QuirogaNo ratings yet
- Your Journey To The Basics Of Quantum Realm Volume II: Your Journey to The Basics Of Quantum Realm, #2From EverandYour Journey To The Basics Of Quantum Realm Volume II: Your Journey to The Basics Of Quantum Realm, #2Rating: 5 out of 5 stars5/5 (1)
- SuperconductivityDocument7 pagesSuperconductivityGopinathan MNo ratings yet
- EC Analog-ElectronicDocument77 pagesEC Analog-ElectronicGopinathan MNo ratings yet
- India Map in Tamil HDDocument5 pagesIndia Map in Tamil HDGopinathan M0% (1)
- 8051 Add Sub ProgramDocument4 pages8051 Add Sub ProgramGopinathan MNo ratings yet
- Nernst EquationDocument8 pagesNernst EquationGopinathan MNo ratings yet
- GenerelDocument11 pagesGenerelGopinathan MNo ratings yet
- CPH 52 I - Iii UnitDocument1 pageCPH 52 I - Iii UnitGopinathan MNo ratings yet
- 2019Document15 pages2019Gopinathan MNo ratings yet
- SpectrumSeries ConceptDocument4 pagesSpectrumSeries ConceptGopinathan MNo ratings yet
- Sri Bharathi Women'S Arts & Science CollegeDocument4 pagesSri Bharathi Women'S Arts & Science CollegeGopinathan MNo ratings yet
- 2018Document15 pages2018Gopinathan MNo ratings yet
- Block GKDocument90 pagesBlock GKGopinathan MNo ratings yet
- BSPH 55 AstrophysicsDocument1 pageBSPH 55 AstrophysicsGopinathan MNo ratings yet
- 10th English Guide - Unit 1 by Loyola PublicationsDocument89 pages10th English Guide - Unit 1 by Loyola PublicationsGopinathan M67% (3)
- CPH 52 Full SyllabusDocument1 pageCPH 52 Full SyllabusGopinathan MNo ratings yet
- A Study of Education On Power TransformersDocument6 pagesA Study of Education On Power TransformersGopinathan MNo ratings yet
- CPH 11Document1 pageCPH 11Gopinathan MNo ratings yet
- Sim Rim Code ConversionDocument23 pagesSim Rim Code ConversionGopinathan MNo ratings yet
- BPH 51 OpticsDocument1 pageBPH 51 OpticsGopinathan MNo ratings yet
- Sri Bharathi Women'S Arts & Science College, Kunnathur, Arni - 632 314Document1 pageSri Bharathi Women'S Arts & Science College, Kunnathur, Arni - 632 314Gopinathan MNo ratings yet
- Sri Bharathi Women'S Arts & Science College Kunnathur, Arni - 632 314Document1 pageSri Bharathi Women'S Arts & Science College Kunnathur, Arni - 632 314Gopinathan MNo ratings yet
- Thiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniDocument10 pagesThiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniGopinathan MNo ratings yet
- BPH 51 OpticsDocument1 pageBPH 51 OpticsGopinathan MNo ratings yet
- Cis TransDocument18 pagesCis TransGopinathan MNo ratings yet
- Thiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniDocument10 pagesThiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniGopinathan MNo ratings yet
- Thiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniDocument10 pagesThiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniGopinathan MNo ratings yet
- 4 Measuring Distances in Space PDFDocument6 pages4 Measuring Distances in Space PDFGopinathan MNo ratings yet
- Thiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniDocument10 pagesThiruvalluvar University: 229-Sri Bharathi Women'S Arts & Science College, Kunnathur-ArniGopinathan MNo ratings yet
- Thiruvalluvar University Astrophysics Internal Assessment QuestionsDocument10 pagesThiruvalluvar University Astrophysics Internal Assessment QuestionsGopinathan MNo ratings yet
- 1.adc DacDocument35 pages1.adc DacGopinathan MNo ratings yet
- ME320 Professor John M. Cimbala: Water Draining From A TankDocument6 pagesME320 Professor John M. Cimbala: Water Draining From A TankShohag HossainNo ratings yet
- Tube Wall Delamination Tubular GC3N4Document16 pagesTube Wall Delamination Tubular GC3N4Nghiêm Thái VĩnhNo ratings yet
- ICDA - MX Line - MRPL - Multiphase Flow Modeling Report Draft 1.0Document16 pagesICDA - MX Line - MRPL - Multiphase Flow Modeling Report Draft 1.0Anonymous AtAGVssJNo ratings yet
- Problem Set No. 3 Thermodynamic Analysis - 2016 v2Document2 pagesProblem Set No. 3 Thermodynamic Analysis - 2016 v2Emrico Luiz PerezNo ratings yet
- Atomic and Molecular Structure ExplainedDocument20 pagesAtomic and Molecular Structure Explainedke.No ratings yet
- Effects of Viscosity of Fluids On Centrifugal Pump Performance and Flow Pattern in The Impeller PDFDocument6 pagesEffects of Viscosity of Fluids On Centrifugal Pump Performance and Flow Pattern in The Impeller PDFLAlvesNo ratings yet
- Steam Bench: ObjectiveDocument6 pagesSteam Bench: Objectiveحسين عمريNo ratings yet
- Pure SubstanceDocument40 pagesPure Substanceamit rajNo ratings yet
- Lesson 3 Pascals PrincipleDocument25 pagesLesson 3 Pascals PrincipleJojimar JulianNo ratings yet
- CrystallizationDocument7 pagesCrystallizationKhaqan AminNo ratings yet
- Chemistry Microproject 1Document12 pagesChemistry Microproject 1Aachal GulhaneNo ratings yet
- Uni Refrigeration System GuideDocument24 pagesUni Refrigeration System Guideminesh.manu6531No ratings yet
- Lecture 16Document11 pagesLecture 16vamsikrishna14No ratings yet
- CET - Module 6Document90 pagesCET - Module 6Ashna GautamNo ratings yet
- Evaporation 2Document18 pagesEvaporation 2Kuma Gloria100% (1)
- Automatic Unloading SystemDocument3 pagesAutomatic Unloading Systemnirmalgupte22No ratings yet
- Chemical Bonding & ElectronegativityDocument4 pagesChemical Bonding & ElectronegativityGwynethh EreseNo ratings yet
- Akash PDFDocument23 pagesAkash PDFAditya KumarNo ratings yet
- D 42. 1053 Kmol/hr B 157. 8947 Kmol/hrDocument2 pagesD 42. 1053 Kmol/hr B 157. 8947 Kmol/hrFran LeeNo ratings yet
- Natural Gas Conversion GuideDocument52 pagesNatural Gas Conversion GuidehonabbieNo ratings yet
- Distillation Equilibrium Problems and Flash CalculationsDocument2 pagesDistillation Equilibrium Problems and Flash CalculationsRosario Zambrano CoronaNo ratings yet
- Viscosity and Consistency Properties of FoodsDocument2 pagesViscosity and Consistency Properties of FoodsAnabelle SabanganNo ratings yet
- Finding Equivalent Resistance in a NetworkDocument20 pagesFinding Equivalent Resistance in a NetworktharinduNo ratings yet
- Subsea Multiphase Compression - From Technology Development To Offshore ...Document35 pagesSubsea Multiphase Compression - From Technology Development To Offshore ...ChadNo ratings yet
- Xample: Internal Incompressible Viscous FlowDocument2 pagesXample: Internal Incompressible Viscous Flowวีรินทร์ ขนิษดาNo ratings yet
- Example 4 Operating Conditions of VesselDocument2 pagesExample 4 Operating Conditions of VesselRindangNo ratings yet
- Characterization of Porous Titanium Dioxide Nanorods-2Document4 pagesCharacterization of Porous Titanium Dioxide Nanorods-2Kshitij RautNo ratings yet
- LAB2 TechnicalReport 19939889Document18 pagesLAB2 TechnicalReport 19939889tichgicNo ratings yet