You might also like
- Dwnload Full Introduction To Quantum Mechanics 3rd Edition Griffiths Solutions Manual PDFDocument20 pagesDwnload Full Introduction To Quantum Mechanics 3rd Edition Griffiths Solutions Manual PDFChristopherNortonwqnmc80% (15)
- Introduction To Nuclear and Particle Physics by V. K. Mittal R. C. Verma S. C. Gupta (Mittal, V. K. Verma, R. C. Gupta, S. C.) PDFDocument438 pagesIntroduction To Nuclear and Particle Physics by V. K. Mittal R. C. Verma S. C. Gupta (Mittal, V. K. Verma, R. C. Gupta, S. C.) PDFx75% (4)
- Science Atoms PDFDocument5 pagesScience Atoms PDFQuennee Ronquillo EscobilloNo ratings yet
- Types of Vibrations in IR Spectroscopy - 07Document27 pagesTypes of Vibrations in IR Spectroscopy - 07آ ئمہNo ratings yet
- Semi Conductor 1Document25 pagesSemi Conductor 1rayina raka mulyanaNo ratings yet
- Cohrence in CinemaDocument8 pagesCohrence in CinemadliyaxNo ratings yet
- Crystals 11 01109Document8 pagesCrystals 11 01109hamza bakkaliNo ratings yet
- Photoinduced Electron Transfer at MoleculeDocument20 pagesPhotoinduced Electron Transfer at MoleculeSumit KumarNo ratings yet
- Double Layer Theory312Document4 pagesDouble Layer Theory312Manal WehbaNo ratings yet
- APL GuntherDocument4 pagesAPL GuntherLiviu BadeaNo ratings yet
- Molecular Modeling of Electron TappingDocument9 pagesMolecular Modeling of Electron TappingJINEETH JJOSEPHNo ratings yet
- Actinide Ground-State Properties: Heoretical PredictionsDocument24 pagesActinide Ground-State Properties: Heoretical PredictionsarchanaphysicstNo ratings yet
- Why Tight-Binding Theory?: Walter A. HarrisonDocument5 pagesWhy Tight-Binding Theory?: Walter A. HarrisonzoehdiismailNo ratings yet
- Becke, 1990. A Simple Measure of Electron Localization in Atomic and Molecular SystemsDocument8 pagesBecke, 1990. A Simple Measure of Electron Localization in Atomic and Molecular SystemsUriel Rangel PenaNo ratings yet
- Energy & Environmental Science: CommunicationDocument6 pagesEnergy & Environmental Science: CommunicationBhabani Sankar SwainNo ratings yet
- Cheng 2018Document6 pagesCheng 2018fouad alimdarNo ratings yet
- 2015 Frustration of Negative Capacitance in AlO, BTOBilayer Structure - YJ Kim (Sicentific Reports)Document11 pages2015 Frustration of Negative Capacitance in AlO, BTOBilayer Structure - YJ Kim (Sicentific Reports)杨珂同No ratings yet
- The Pseudopotential PanaceaDocument9 pagesThe Pseudopotential PanaceaMedo MedicNo ratings yet
- Computational Method To Estimate Single Event Upset Rates in An Accelerator EnvironmentDocument18 pagesComputational Method To Estimate Single Event Upset Rates in An Accelerator Environmentbububu66No ratings yet
- Boukamp-2020-Journal of Physics EnergyDocument24 pagesBoukamp-2020-Journal of Physics EnergyJayveer JoshiNo ratings yet
- Narducci 2012Document7 pagesNarducci 2012Asep Ridwan NugrahaNo ratings yet
- Journal: Origin of The Potential Drop Over The Deposit During Electrophoretic DepositionDocument6 pagesJournal: Origin of The Potential Drop Over The Deposit During Electrophoretic DepositionMario Misael Machado LòpezNo ratings yet
- Boukamp 2020 J. Phys. Energy 2 042001Document25 pagesBoukamp 2020 J. Phys. Energy 2 042001Jayveer JoshiNo ratings yet
- The Electron and The Holographic Mass Solution A) : Physics Essays June 2019Document9 pagesThe Electron and The Holographic Mass Solution A) : Physics Essays June 2019AmiNo ratings yet
- Relaxation and Edge Reconstruction in Integer Quantum Hall SystemsDocument15 pagesRelaxation and Edge Reconstruction in Integer Quantum Hall SystemsDustisNo ratings yet
- Artigo JMMDocument11 pagesArtigo JMMMaurício FalleirosNo ratings yet
- Chemical Physics Letters: Arthur J. NozikDocument9 pagesChemical Physics Letters: Arthur J. NozikPedroLozanoNo ratings yet
- 2011 MPSD Qcm+et - Petersen PRLDocument5 pages2011 MPSD Qcm+et - Petersen PRLnickydean83No ratings yet
- Analytic Model of The Cathode Region of A Short Glow Discharge in Light GasesDocument16 pagesAnalytic Model of The Cathode Region of A Short Glow Discharge in Light GasesLeonardoNo ratings yet
- Material Properties and Device Physics Basic To PhotovoltaicsDocument55 pagesMaterial Properties and Device Physics Basic To Photovoltaics呂宗霖No ratings yet
- Introduction To PhotovoltaicDocument11 pagesIntroduction To PhotovoltaicGautam Kumar DeepakNo ratings yet
- Chiral Tunneling and The Klein Paradox in GrapheneDocument15 pagesChiral Tunneling and The Klein Paradox in GrapheneGhidic VladislavNo ratings yet
- Insulating To Metallic TransitionDocument8 pagesInsulating To Metallic TransitionSandeepKumarSinghNo ratings yet
- 10 1002@adfm 201910461Document8 pages10 1002@adfm 201910461Abdelkader NadjemNo ratings yet
- Topological InsulatorsDocument9 pagesTopological InsulatorsRiturajNo ratings yet
- Orbital-Selective Charge-Density Wave in Tate: ArticleDocument7 pagesOrbital-Selective Charge-Density Wave in Tate: ArticleangelomalachiasNo ratings yet
- Spin ValleyDocument7 pagesSpin ValleyLakpa TamangNo ratings yet
- ElectrophoresisDocument31 pagesElectrophoresisDr. Rabia NazNo ratings yet
- Ground State Energy of Hydrogen-Like Ions in Quantum PlasmasDocument13 pagesGround State Energy of Hydrogen-Like Ions in Quantum PlasmasJit SarkarNo ratings yet
- PhysRevA 14 2061Document3 pagesPhysRevA 14 2061Meidk LoreNo ratings yet
- PhysRevA 98 043801Document24 pagesPhysRevA 98 043801Geziane Santos PereiraNo ratings yet
- PhysRevLett 112 113002Document5 pagesPhysRevLett 112 113002liopoiNo ratings yet
- A Spin-Coherent Semiconductor Photo-Detector For Quantum CommunicationDocument7 pagesA Spin-Coherent Semiconductor Photo-Detector For Quantum CommunicationkamasanisNo ratings yet
- Energy-Scale Cascade and Correspondence Between The Mott and Kondo Lattice PhysicsDocument7 pagesEnergy-Scale Cascade and Correspondence Between The Mott and Kondo Lattice PhysicsXuan LiuNo ratings yet
- Parameters Calculation and Model IntegrationDocument10 pagesParameters Calculation and Model IntegrationRafael Becker MacielNo ratings yet
- Photonic Topological Insulators A Beginners Introduction Electromagnetic PerspectivesDocument13 pagesPhotonic Topological Insulators A Beginners Introduction Electromagnetic PerspectivesabduNo ratings yet
- 1quantumchemical Electronegativity and Hardness IndicesDocument16 pages1quantumchemical Electronegativity and Hardness IndicesSupreeth PrasadNo ratings yet
- Quantum Information Processing Using Molecular Nanomagnets As Qubits (Angewandte Chemie, Vol. 47, Issue 42) (2008)Document3 pagesQuantum Information Processing Using Molecular Nanomagnets As Qubits (Angewandte Chemie, Vol. 47, Issue 42) (2008)sepot24093No ratings yet
- Impedence SpectrosDocument110 pagesImpedence SpectrosManu PriyaNo ratings yet
- Physical Revie% December Atomic: FirstDocument3 pagesPhysical Revie% December Atomic: FirstMeidk LoreNo ratings yet
- Single Wall Carbon Nanotubes/Polypyrrole Composite Thin Film Electrodes: Investigation of Interfacial Ion Exchange BehaviorDocument14 pagesSingle Wall Carbon Nanotubes/Polypyrrole Composite Thin Film Electrodes: Investigation of Interfacial Ion Exchange BehaviormiguelNo ratings yet
- Tunneling-Assisted Poole-Frenkel Conduction Mechanism in HfO2 Thin FilmsDocument7 pagesTunneling-Assisted Poole-Frenkel Conduction Mechanism in HfO2 Thin FilmsDiego Carranza CelisNo ratings yet
- Jamnik 1999Document6 pagesJamnik 1999Ananth BalakrishnanNo ratings yet
- The Structure of Atomic NucleusDocument7 pagesThe Structure of Atomic NucleusDezso Sarkadi100% (1)
- Nature 08293Document5 pagesNature 08293pyczy45678No ratings yet
- Paper 2Document8 pagesPaper 2Alexander AndradeNo ratings yet
- Orientational Ordering and Electron-Phonon Interaction in K3C60 SuperconductorDocument8 pagesOrientational Ordering and Electron-Phonon Interaction in K3C60 SuperconductorEXITOFORTUNANo ratings yet
- Conducting Polymers: Carly Anderson and Emily Davidson April 23, 2013Document29 pagesConducting Polymers: Carly Anderson and Emily Davidson April 23, 2013AdityaNo ratings yet
- The Transition To The Metallic State in Polycrystalline N-Type Doped ZnODocument9 pagesThe Transition To The Metallic State in Polycrystalline N-Type Doped ZnORachid MakhloufiNo ratings yet
- Physica B: Peng-Fei Cheng, Jiang Song, Sheng-Tao Li, Hui WangDocument5 pagesPhysica B: Peng-Fei Cheng, Jiang Song, Sheng-Tao Li, Hui Wangfouad alimdarNo ratings yet
- Cesium Telluride Photocathodes: Journal of Applied Physics July 1995Document10 pagesCesium Telluride Photocathodes: Journal of Applied Physics July 1995sandeepNo ratings yet
- Observation of Dirac Charge Density Waves in Bi Te SeDocument8 pagesObservation of Dirac Charge Density Waves in Bi Te SePhysics FizicaNo ratings yet
- Mitigation of Electron Cloud Effects in The FCC-ee ColliderDocument11 pagesMitigation of Electron Cloud Effects in The FCC-ee ColliderFatihNo ratings yet
- Steady Electric Fields and Currents: Elementary Electromagnetic TheoryFrom EverandSteady Electric Fields and Currents: Elementary Electromagnetic TheoryNo ratings yet
- The Physics PGDocument23 pagesThe Physics PGAurobind DasNo ratings yet
- Physics - Zero Point EnergyDocument28 pagesPhysics - Zero Point Energymakaliza2005No ratings yet
- Particle Wave DualityDocument24 pagesParticle Wave DualityIsrael PopeNo ratings yet
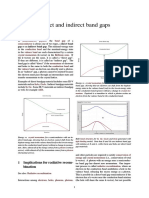
- Direct and Indirect Band GapsDocument4 pagesDirect and Indirect Band GapsSai Swetha KVNo ratings yet
- MSC Physics SyllabusDocument47 pagesMSC Physics SyllabusAneez KoyattyNo ratings yet
- Challenges and Opportunities in Quantum Machine LearningDocument14 pagesChallenges and Opportunities in Quantum Machine LearningDanny XuNo ratings yet
- Waves and Particles in Light and MatterDocument8 pagesWaves and Particles in Light and MatterminhankyawNo ratings yet
- Physical Chemistry (III)Document32 pagesPhysical Chemistry (III)GraceNo ratings yet
- Bioplasma Concept of ConsciousnessDocument11 pagesBioplasma Concept of ConsciousnessJeremy Oneal100% (2)
- LightDocument29 pagesLightMark Louis SantiagoNo ratings yet
- C1 ErroriDocument164 pagesC1 ErroriVitalie LunguNo ratings yet
- Engineering Physics Syllabus 2018 OnwardsDocument2 pagesEngineering Physics Syllabus 2018 OnwardsRathinaKumarNo ratings yet
- String Theory: A Field Theory Perspective: Harold ErbinDocument350 pagesString Theory: A Field Theory Perspective: Harold ErbineliotthoarauNo ratings yet
- Griffiths Problems 02.49Document3 pagesGriffiths Problems 02.49Ema MaharaniNo ratings yet
- Raman Spectroscopy: Principles and Applications: M. K. Panigrahi Department of Geology & GeophysicsDocument29 pagesRaman Spectroscopy: Principles and Applications: M. K. Panigrahi Department of Geology & GeophysicsPradeep SijeriyaNo ratings yet
- Sri Chaitanya IIT Academy., India.: KEY SheetDocument6 pagesSri Chaitanya IIT Academy., India.: KEY SheetO SNo ratings yet
- Band Theory-1 PDFDocument57 pagesBand Theory-1 PDFambidextrousalienNo ratings yet
- CHEMISTRY TOPIC 3 TrendsDocument3 pagesCHEMISTRY TOPIC 3 TrendsAisy BalalaNo ratings yet
- Instrukcja PS1 cw4Document22 pagesInstrukcja PS1 cw4verenichdanielfiNo ratings yet
- GENERAL CHEMISTRY 1 Quarter 2 Module 1Document14 pagesGENERAL CHEMISTRY 1 Quarter 2 Module 1lkNo ratings yet
- Tao Et Al. (2010)Document5 pagesTao Et Al. (2010)mateus_rocha0% (1)
- Hydrogen-Like Atoms: Energy LevelDocument13 pagesHydrogen-Like Atoms: Energy LevelJohn DoeNo ratings yet
- Quantum States of Electrons in AtomsDocument5 pagesQuantum States of Electrons in Atomskhy82No ratings yet
- PHYS532 - Quantum Mechanics II Problem Set 3 DueDocument1 pagePHYS532 - Quantum Mechanics II Problem Set 3 DuewaskhezNo ratings yet
- Joey Sulpizio - Quantum Transport in One-Dimensional Nanostructures PDFDocument171 pagesJoey Sulpizio - Quantum Transport in One-Dimensional Nanostructures PDFsethupathiNo ratings yet
- 1 of 39 © Boardworks LTD 2009Document19 pages1 of 39 © Boardworks LTD 2009krisnuNo ratings yet