You might also like
- Cardiovascular System Dr. Eman Badr 2020Document182 pagesCardiovascular System Dr. Eman Badr 2020Amina DinarNo ratings yet
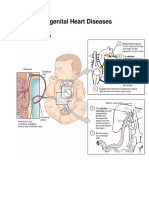
- Conginital Heart DiseaseDocument19 pagesConginital Heart DiseaseSanthosh.S.UNo ratings yet
- Ventricular Septal DefectDocument6 pagesVentricular Septal DefectYannah Mae EspineliNo ratings yet
- Lapkas Dian and DamboDocument34 pagesLapkas Dian and DamboDian Primadia PutriNo ratings yet
- Tetralogy of FallotDocument31 pagesTetralogy of FallotAnditha Namira RS100% (1)
- Congenital Heart Disease Is One or More Problems With The Heart's Structure ThatDocument37 pagesCongenital Heart Disease Is One or More Problems With The Heart's Structure Thatneelimawanker chinnariNo ratings yet
- Nursing Care Plan For Acyanotic Heart DiseaseDocument55 pagesNursing Care Plan For Acyanotic Heart DiseaseDeepikaxena John79% (14)
- Transposition of Great Artery: 1. AbstractDocument7 pagesTransposition of Great Artery: 1. AbstractA. Fajar AprianiNo ratings yet
- Atrial Septal DefectDocument5 pagesAtrial Septal DefectalmorsNo ratings yet
- Herlina DimiatiDocument25 pagesHerlina DimiatikholilgantengNo ratings yet
- Atrial Septal DefectDocument7 pagesAtrial Septal DefectRis Amy AyaNo ratings yet
- Atrial Septal DefectDocument3 pagesAtrial Septal Defectktin17No ratings yet
- Cardiovascular Dysfunction in ChildrenDocument15 pagesCardiovascular Dysfunction in ChildrenJhasseryne Orias SanchezNo ratings yet
- Understanding Tetralogy of Fallot: Causes, Symptoms and TreatmentDocument7 pagesUnderstanding Tetralogy of Fallot: Causes, Symptoms and TreatmentJoanna Bee Rose MagyawiNo ratings yet
- Congenital Heart DiseaseDocument93 pagesCongenital Heart DiseaseManjunatha HR100% (1)
- Cardio Vascular DiseasesDocument34 pagesCardio Vascular DiseasesSam ParkNo ratings yet
- What Causes Increased Pulmonary Blood FlowDocument11 pagesWhat Causes Increased Pulmonary Blood FlowJc Pauline PabustanNo ratings yet
- Congenital Heart Disease - Cynotic AcynoticDocument34 pagesCongenital Heart Disease - Cynotic Acynoticvruttika parmarNo ratings yet
- CVS - Session 3 - CHDDocument3 pagesCVS - Session 3 - CHDkotecha.rheaNo ratings yet
- Congenital Heart DiseasesDocument55 pagesCongenital Heart DiseasesRajaNo ratings yet
- Minggu 2 LP ASDDocument15 pagesMinggu 2 LP ASDMuhammad PanduNo ratings yet
- Tricuspid Atresia Heart Defect ExplainedDocument5 pagesTricuspid Atresia Heart Defect ExplainedCassyDelaRosaNo ratings yet
- Congenital Heart DiseasesDocument55 pagesCongenital Heart DiseasesFaheem BasharatNo ratings yet
- Case Report: Atrial Septal Defect and Ventricular Septal DefectDocument28 pagesCase Report: Atrial Septal Defect and Ventricular Septal DefectDian Primadia PutriNo ratings yet
- Congenital Heart Diseases, A Simple Guide to these Medical ConditionsFrom EverandCongenital Heart Diseases, A Simple Guide to these Medical ConditionsNo ratings yet
- Congenital Heart DefectDocument10 pagesCongenital Heart DefectShubhamNo ratings yet
- Cardio Vascular DisordersDocument62 pagesCardio Vascular DisordersUday Kumar100% (1)
- Materi TM 1Document3 pagesMateri TM 1leny cahyani putriNo ratings yet
- What Is Tetralogy of Fallot?: Congenital Heart DefectDocument3 pagesWhat Is Tetralogy of Fallot?: Congenital Heart DefectGlenn Asuncion PagaduanNo ratings yet
- Tetralogy of FallotDocument5 pagesTetralogy of FallotCharity OaniaNo ratings yet
- Decomp Cordis (Fais & Dalilah)Document10 pagesDecomp Cordis (Fais & Dalilah)Dalilah VitrianaNo ratings yet
- TiKa TaKa PediatricsDocument86 pagesTiKa TaKa PediatricsEmad MerganNo ratings yet
- Cardiac Disorders in People With Down's Syndrome: The Normal HeartDocument5 pagesCardiac Disorders in People With Down's Syndrome: The Normal HeartGeorgiana RadoslavNo ratings yet
- Tetralogy of FallotDocument7 pagesTetralogy of FallotAshok Kumar JangirNo ratings yet
- Congenital Heart DiseaseDocument31 pagesCongenital Heart DiseaseSandeep ChaudharyNo ratings yet
- Tetralogy of FallotDocument28 pagesTetralogy of FallotconcozNo ratings yet
- Tetralogy of FallotDocument38 pagesTetralogy of FallotJohn Paul MedalloNo ratings yet
- Aruna Ramesh-Emergency...Document6 pagesAruna Ramesh-Emergency...Aishu BNo ratings yet
- TYPES OF HEART DEFECTSDocument11 pagesTYPES OF HEART DEFECTSJuliet Marie MiomioNo ratings yet
- Congenital Heart Disease GuideDocument89 pagesCongenital Heart Disease GuideMathew JosephNo ratings yet
- Introtroduction and Classification of Heart DiseasesDocument30 pagesIntrotroduction and Classification of Heart DiseasesNITHA KNo ratings yet
- Congenital Heart Diseases.Document86 pagesCongenital Heart Diseases.Salman KhanNo ratings yet
- How The Heart Works: Septum Lungs OxygenDocument22 pagesHow The Heart Works: Septum Lungs OxygenSuzan IbrahimNo ratings yet
- Acyanotic Congenital Heart DiseaseDocument7 pagesAcyanotic Congenital Heart DiseaseSam Raj100% (1)
- Lecture 1 Heart OverviewDocument20 pagesLecture 1 Heart OverviewOsama MalikNo ratings yet
- Congenital Heart Disease EtiologyDocument8 pagesCongenital Heart Disease EtiologykudzaimuregidubeNo ratings yet
- Congenital Heart DiseaseDocument12 pagesCongenital Heart Diseaserakanootousan100% (1)
- Congenital Heart Disease 1223958712449685 9Document58 pagesCongenital Heart Disease 1223958712449685 9RinaNo ratings yet
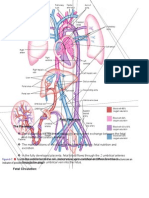
- Fetal Perfusion/Congenital Heart DefectsDocument13 pagesFetal Perfusion/Congenital Heart DefectsMatthew RyanNo ratings yet
- What is Tetralogy of FallotDocument2 pagesWhat is Tetralogy of FallotJamaica Cruz San Pedro100% (1)
- Risk Factors and Disease Progression in Tetralogy of FallotDocument5 pagesRisk Factors and Disease Progression in Tetralogy of FallotJoanna Bee Rose MagyawiNo ratings yet
- Congenital Heart Disease (2011)Document17 pagesCongenital Heart Disease (2011)drheay100% (1)
- Tetralogy of FallotDocument3 pagesTetralogy of FallotMiLody NajeNo ratings yet
- Tricuspid AtresiaDocument14 pagesTricuspid AtresiaPretty AjursNo ratings yet
- PJB Pada Dewasa AASDocument54 pagesPJB Pada Dewasa AAS1e23e2ewNo ratings yet
- Congenital Heart DiseasesDocument12 pagesCongenital Heart DiseasesFrinkaWijayaNo ratings yet
- Tumbang KabehDocument57 pagesTumbang KabehArif WicaksanaNo ratings yet
- Reverse and Prevent Heart Disease: Natural Ways to Stop and Prevent Heart Disease, Using Plant-Based Oil-Free Diets (Cure Congestive Heart Failure)From EverandReverse and Prevent Heart Disease: Natural Ways to Stop and Prevent Heart Disease, Using Plant-Based Oil-Free Diets (Cure Congestive Heart Failure)No ratings yet
- Critique Paper01 - DabloDocument2 pagesCritique Paper01 - DabloFheevNo ratings yet
- FULL Download Ebook PDF Fundamentals of Health Psychology 2nd Edition PDF EbookDocument41 pagesFULL Download Ebook PDF Fundamentals of Health Psychology 2nd Edition PDF Ebookjennifer.lawver532100% (31)
- Hortatory Exposition: Anang Tri Olivandy Baiq Wimandari Safitri Raissa Putri NabellaDocument32 pagesHortatory Exposition: Anang Tri Olivandy Baiq Wimandari Safitri Raissa Putri NabellaAnnisa ArsyNo ratings yet
- J.S Case Study 1 PDFDocument2 pagesJ.S Case Study 1 PDFania ojedaNo ratings yet
- Nursing Care Plan On Platelet DisordersDocument8 pagesNursing Care Plan On Platelet DisordersbhavanaNo ratings yet
- Download ebook Electromyography And Neuromuscular Disorders Clinical Electrophysiologic Ultrasound Correlations Pdf full chapter pdfDocument68 pagesDownload ebook Electromyography And Neuromuscular Disorders Clinical Electrophysiologic Ultrasound Correlations Pdf full chapter pdfhelen.reid678100% (23)
- Community Mental Health Framework For Adults and Older AdultsDocument22 pagesCommunity Mental Health Framework For Adults and Older AdultsLuigi LuiNo ratings yet
- Oseltamivir Indication, Dosage, Side Effect, Precaution MIMS IndonesiaDocument1 pageOseltamivir Indication, Dosage, Side Effect, Precaution MIMS Indonesiaintan alvinnNo ratings yet
- NEURO VITAL SIGNS ASSESSMENT Procedure and ChecklistDocument12 pagesNEURO VITAL SIGNS ASSESSMENT Procedure and ChecklistMemer-alasadNo ratings yet
- Part 5 Emergency Response PlanDocument23 pagesPart 5 Emergency Response Planalex.kollosovNo ratings yet
- Comparative Single-Dose Pharmacokinetics of Amantadine Hydrochloride and Rimantadine Hydrochloride in Elderly AdultsDocument6 pagesComparative Single-Dose Pharmacokinetics of Amantadine Hydrochloride and Rimantadine Hydrochloride in Elderly AdultsLety LagunaNo ratings yet
- Croup: Assessment and ManagementDocument3 pagesCroup: Assessment and Managementsandra magdalenaNo ratings yet
- Research in Public Health by Prof DR Rwamakuba ZephanieDocument120 pagesResearch in Public Health by Prof DR Rwamakuba ZephanieDr Zephanie RwamakubaNo ratings yet
- Juvenile Idiopathic ArthritisDocument19 pagesJuvenile Idiopathic ArthritisMobin Ur Rehman KhanNo ratings yet
- 5 Common Environmental Hazards in The Workplace: Skip To Main ContentDocument48 pages5 Common Environmental Hazards in The Workplace: Skip To Main ContentElvie Rodado GubalaneNo ratings yet
- Cysts of The JawsDocument25 pagesCysts of The Jawsluna zeidNo ratings yet
- IoT Deployable Lightweight Deep Learning Application For COVID-19 Detection With Lung Diseases Using RaspberryPiDocument6 pagesIoT Deployable Lightweight Deep Learning Application For COVID-19 Detection With Lung Diseases Using RaspberryPiGigin GinanjarNo ratings yet
- Heart Disease in PregnancyDocument56 pagesHeart Disease in PregnancyyandraNo ratings yet
- Ejr D 22 02490Document30 pagesEjr D 22 02490snehaNo ratings yet
- Family Centered CareDocument7 pagesFamily Centered CareSirisha ChelvaNo ratings yet
- DRRR Module 4 5Document4 pagesDRRR Module 4 5kellan lyfeNo ratings yet
- Geriatric Physical AssessmentDocument6 pagesGeriatric Physical AssessmentKrizzle Mae NeypesNo ratings yet
- Viruses Bacteria Protists and FungiDocument63 pagesViruses Bacteria Protists and Fungim umair zahirNo ratings yet
- SCORE Project Form Internal Medicine DepartmentDocument7 pagesSCORE Project Form Internal Medicine DepartmentGeorge CobuzNo ratings yet
- Fluid Consumption, Hydration Status and Its Associated Factors - A Cross Sectional Study Among Medical Students in Palembang, IndonesiaDocument36 pagesFluid Consumption, Hydration Status and Its Associated Factors - A Cross Sectional Study Among Medical Students in Palembang, IndonesiaDwi Lisa Nur'ainiNo ratings yet
- MEGALOBLASTIC ANAEMIA - pptxsdd.pptx.4Document30 pagesMEGALOBLASTIC ANAEMIA - pptxsdd.pptx.4Hiba MohammedNo ratings yet
- Drug Study: Amoxicillin Mechanism and Nursing ResponsibilitiesDocument3 pagesDrug Study: Amoxicillin Mechanism and Nursing ResponsibilitiesKrzia TehNo ratings yet
- Case Study On ManiaDocument30 pagesCase Study On ManiaIOM BNS75% (8)
- Life Cycle of Taenia Saginata and T. SoliumDocument1 pageLife Cycle of Taenia Saginata and T. SoliumPrabin ShakyaNo ratings yet
- How-Do-I-Treat-My-Cough-At-Night-Parkinson PatientDocument1 pageHow-Do-I-Treat-My-Cough-At-Night-Parkinson PatientMuhammad BabarNo ratings yet