You might also like
- Treatise on the Anatomy and Physiology of the Mucous Membranes: With Illustrative Pathological ObservationsFrom EverandTreatise on the Anatomy and Physiology of the Mucous Membranes: With Illustrative Pathological ObservationsNo ratings yet
- Mucociliary ClearanceDocument9 pagesMucociliary Clearancerbatjun576No ratings yet
- Fibrosis QuisticaDocument6 pagesFibrosis QuisticaFernanda Del ValleNo ratings yet
- 2.BE PathogenesisDocument7 pages2.BE PathogenesisJaya Semara PutraNo ratings yet
- Theme1AppliedLecture2 BIO1A03Fall2022Document27 pagesTheme1AppliedLecture2 BIO1A03Fall2022proplayer910No ratings yet
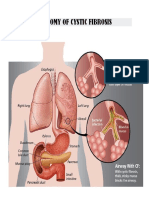
- Anatomy of Cystic FibrosisDocument5 pagesAnatomy of Cystic FibrosislazylawatudentNo ratings yet
- Fimmu 11 01438Document17 pagesFimmu 11 01438anu deepNo ratings yet
- Abnormal Chest FindingsDocument5 pagesAbnormal Chest FindingsAbishek Prince100% (1)
- Cystic FibrosisDocument13 pagesCystic FibrosisAnkita guptaNo ratings yet
- BronchiectasisDocument4 pagesBronchiectasisCitrusNo ratings yet
- Chapter44 MiddletonAllergy9eDocument13 pagesChapter44 MiddletonAllergy9eIkon ikonicNo ratings yet
- Emerging Gene Therapies For Cystic Fibrosis: Expert Review of Respiratory MedicineDocument18 pagesEmerging Gene Therapies For Cystic Fibrosis: Expert Review of Respiratory MedicineIzabelly OliveiraNo ratings yet
- BronchiectasisDocument9 pagesBronchiectasisIS99057No ratings yet
- Chronic Obstructive Pulmonary Disease: Iman Galal, MDDocument60 pagesChronic Obstructive Pulmonary Disease: Iman Galal, MDYan Sheng HoNo ratings yet
- Harrisons Principles of Internal Medicine, 19th EditionDocument8 pagesHarrisons Principles of Internal Medicine, 19th EditionTALBIYAH SABDAH RIZAN TAUPIQ -No ratings yet
- Interstitial Lung DiseaseDocument15 pagesInterstitial Lung DiseaseLea MonzerNo ratings yet
- Di¡erential Adaptation of Microbial Pathogens To Airways of Patients With Cystic Brosis and Chronic Obstructive Pulmonary DiseaseDocument24 pagesDi¡erential Adaptation of Microbial Pathogens To Airways of Patients With Cystic Brosis and Chronic Obstructive Pulmonary DiseaseAbd Halim Gazali HNo ratings yet
- AutopsyDocument29 pagesAutopsyAkshita Amit AgarwalNo ratings yet
- Activity Respiratory SystemDocument2 pagesActivity Respiratory SystemBianca CarillaNo ratings yet
- Cystic FibrosisDocument3 pagesCystic FibrosisstanscimagNo ratings yet
- Cystic Fibrosis - Dr. IonescuDocument60 pagesCystic Fibrosis - Dr. IonescuArleen MatincaNo ratings yet
- Empyema - Rol of SurgeonDocument5 pagesEmpyema - Rol of Surgeondantheman123No ratings yet
- Cystic FibrosisDocument31 pagesCystic FibrosisshadabNo ratings yet
- Cystic Fibrosis LectureDocument16 pagesCystic Fibrosis Lecturevani reddyNo ratings yet
- Chronic Obstructive Pulmonary Disease: PathogenesisDocument10 pagesChronic Obstructive Pulmonary Disease: PathogenesisJacob BorongNo ratings yet
- 109 - Obstructive Lung DiseasesDocument8 pages109 - Obstructive Lung DiseasesCharisa Antonette HuelvaNo ratings yet
- Fibrosis Quística y Enfermedad PulmonarDocument9 pagesFibrosis Quística y Enfermedad PulmonarMELERYN CRISTY VELASQUEZ MOSTACERONo ratings yet
- Imaging in Acute Appendicitis: A Review: RK Jain, M Jain, CL Rajak, S Mukherjee, PP Bhattacharyya, MR ShahDocument10 pagesImaging in Acute Appendicitis: A Review: RK Jain, M Jain, CL Rajak, S Mukherjee, PP Bhattacharyya, MR ShahVidini Kusuma AjiNo ratings yet
- Bronchiectasis PathophysiologyDocument4 pagesBronchiectasis PathophysiologyBrunhild BangayanNo ratings yet
- Chapter Title: Pulmonary Disease ManagementDocument5 pagesChapter Title: Pulmonary Disease ManagementCharisa Antonette HuelvaNo ratings yet
- Image Session of Respiratory System IDocument31 pagesImage Session of Respiratory System IZain Ul AbidinNo ratings yet
- Medical Progress: Review ArticleDocument11 pagesMedical Progress: Review ArticlemayaNo ratings yet
- Chronic Obstructive Pulmonary Disease: PathogenesisDocument9 pagesChronic Obstructive Pulmonary Disease: PathogenesisMerienda TimeNo ratings yet
- Medicine (Dr. Pedroza) Copd 14 FEBRUARY 2018Document3 pagesMedicine (Dr. Pedroza) Copd 14 FEBRUARY 2018Seff CausapinNo ratings yet
- Chapter 259. Cystic FibrosisDocument7 pagesChapter 259. Cystic FibrosisAntónio CarvalhoNo ratings yet
- Parasitic Infections of The Lung: A Guide For The Respiratory PhysicianDocument9 pagesParasitic Infections of The Lung: A Guide For The Respiratory Physicianحسام الدين إسماعيلNo ratings yet
- CFTR 2Document8 pagesCFTR 2Linda KurniawanNo ratings yet
- BronchiectasisDocument64 pagesBronchiectasisbrijeshNo ratings yet
- ORENSTEIN 2002 - Cystic Fibrosis - A 2002 UpdateDocument9 pagesORENSTEIN 2002 - Cystic Fibrosis - A 2002 UpdateRafael JustinoNo ratings yet
- Parasitic Infections of The Lung: A Guide For The Respiratory PhysicianDocument9 pagesParasitic Infections of The Lung: A Guide For The Respiratory PhysicianSuria KumarNo ratings yet
- Schematic Diagram OF BronchopneumoniaDocument11 pagesSchematic Diagram OF BronchopneumoniaanGel1115No ratings yet
- The Cystic Fibrosis Transmembrane Conductance Regulator: An Intriguing Protein With Pleitropic FunctionsDocument17 pagesThe Cystic Fibrosis Transmembrane Conductance Regulator: An Intriguing Protein With Pleitropic FunctionsKaren NietoNo ratings yet
- Cystic FibrosisDocument33 pagesCystic FibrosisGreeshma ShajiNo ratings yet
- Etiology: An Irreversible Airway Dilation That Involves The Lung inDocument3 pagesEtiology: An Irreversible Airway Dilation That Involves The Lung inOlli TherineNo ratings yet
- Hamilos 2016. Chronic Rhinosinusitis in Patients With Cystic FibrosisDocument8 pagesHamilos 2016. Chronic Rhinosinusitis in Patients With Cystic FibrosisgulararezendeNo ratings yet
- A Case of PseudochylothoraxDocument3 pagesA Case of PseudochylothoraxdrdodaNo ratings yet
- Resp Tract Epithelium HypoxiaDocument16 pagesResp Tract Epithelium Hypoxiaedgar davilaNo ratings yet
- Silicosis Presenting With Simultaneous Bilateral Spontaneous PneumothoraxDocument3 pagesSilicosis Presenting With Simultaneous Bilateral Spontaneous PneumothoraxSuman RaushanNo ratings yet
- Epithelial Barriers in Allergy and AsthmaDocument11 pagesEpithelial Barriers in Allergy and AsthmaFrancisco Baca DejoNo ratings yet
- Challenges in Pulmonary Fibrosis ? 3: Cystic Lung Disease: Review SeriesDocument11 pagesChallenges in Pulmonary Fibrosis ? 3: Cystic Lung Disease: Review SeriesedelinNo ratings yet
- Fibrosis Quística - NEJM02112023Document15 pagesFibrosis Quística - NEJM02112023Jesus A. Pineda GarciaNo ratings yet
- Cystic Fibrosis PathophysiologyDocument5 pagesCystic Fibrosis PathophysiologyKim Enrico JumarangNo ratings yet
- BRONCHIECTASISDocument40 pagesBRONCHIECTASISImmanuel100% (2)
- Diffuse Lung Disease Caused by Cotton Fibre Inhalation But Distinct From ByssinosisDocument3 pagesDiffuse Lung Disease Caused by Cotton Fibre Inhalation But Distinct From ByssinosisAnonymous h1XAlApsUNo ratings yet
- Non-Cystic Fibrosis Bronchiectasis: Diagnosis and Management in 21st CenturyDocument10 pagesNon-Cystic Fibrosis Bronchiectasis: Diagnosis and Management in 21st CenturyIvan VeriswanNo ratings yet
- BronchiectasisDocument21 pagesBronchiectasisMuhammad Iyhan NuriansyahNo ratings yet
- Pathology of Airway DiseasesDocument42 pagesPathology of Airway DiseasesZijieNo ratings yet
- The Epithelial Sodium Channel (Enac) As A Therapeutic Target For Cystic Fibrosis Lung DiseaseDocument17 pagesThe Epithelial Sodium Channel (Enac) As A Therapeutic Target For Cystic Fibrosis Lung DiseaseNeysa ArdraNo ratings yet
- Fpi Nejm Clincal Practice 2018Document13 pagesFpi Nejm Clincal Practice 2018cjonderedsonNo ratings yet
- Tuberculous Lymphadenitis: History PathogenesisDocument6 pagesTuberculous Lymphadenitis: History PathogenesisMochamad BilalNo ratings yet
- High-Yield NephrologyDocument6 pagesHigh-Yield NephrologyAhmad SobihNo ratings yet
- BIOL 3121 - Lecture 13 - 081116Document91 pagesBIOL 3121 - Lecture 13 - 081116Sara HuebnerNo ratings yet
- Final Project Report Kiran MazumdarDocument31 pagesFinal Project Report Kiran MazumdarPrajot Morajkar100% (3)
- Composite Fish Culture in PondsDocument2 pagesComposite Fish Culture in Pondsvikasdevre1No ratings yet
- Problem SoilsDocument18 pagesProblem SoilsselvakrishnaNo ratings yet
- Dihybrid Punnett SquaresDocument4 pagesDihybrid Punnett SquaresThomas Abich100% (1)
- Cham PowerpointDocument19 pagesCham PowerpointElizabeth GenotivaNo ratings yet
- Social Institutions - Kinship, Family&Marriage PDFDocument47 pagesSocial Institutions - Kinship, Family&Marriage PDFAbhijith KNo ratings yet
- Noncanonical Transnitrosylation Network Contributes To Synapse Loss in Alzheimer's DiseaseDocument18 pagesNoncanonical Transnitrosylation Network Contributes To Synapse Loss in Alzheimer's DiseasedmitworNo ratings yet
- A Double-Strain TM (gp45) Polypeptide Antigen and Its Application in The Serodiadnosis of Equine Infectius AnemiaDocument8 pagesA Double-Strain TM (gp45) Polypeptide Antigen and Its Application in The Serodiadnosis of Equine Infectius AnemiaFredy MoralesNo ratings yet
- Recent DevelopmentsDocument295 pagesRecent DevelopmentsGabriel PinheiroNo ratings yet
- Is A Calorie Really A Calorie - Metabolic Advantage of Low-Carbohydrate DietsDocument6 pagesIs A Calorie Really A Calorie - Metabolic Advantage of Low-Carbohydrate DietsGustavo CastroNo ratings yet
- Grade 5, Science CurriculumDocument3 pagesGrade 5, Science CurriculumJoel C. Yuvienco67% (3)
- Clave Genero Dermanura (Otros) PDFDocument520 pagesClave Genero Dermanura (Otros) PDFJuan D Garcia Peluffo100% (1)
- Chapter 1Document4 pagesChapter 1Nikoleta RudnikNo ratings yet
- USAID BD Handbook Oct 2015 508Document280 pagesUSAID BD Handbook Oct 2015 508vasconcelos1322No ratings yet
- 7 Pearson SquareDocument19 pages7 Pearson Squareapi-235944649No ratings yet
- EuglenidDocument4 pagesEuglenidx456456456xNo ratings yet
- An Examination and Critique of Current Methods To Determine Exercise IntensityDocument28 pagesAn Examination and Critique of Current Methods To Determine Exercise IntensityPabloAñonNo ratings yet
- Reprogramming Microbes To Be Pathogen-Seeking KillersDocument10 pagesReprogramming Microbes To Be Pathogen-Seeking Killerstian luoNo ratings yet
- Form 2 Notes-1Document53 pagesForm 2 Notes-1William NjorogeNo ratings yet
- MoPSE Science Module Volume 1 - FINAL4WEBDocument336 pagesMoPSE Science Module Volume 1 - FINAL4WEBSibusiso Ntomby NkalaNo ratings yet
- Short Note Biology Form 5-Chapter 3 Coordination and ResponseDocument6 pagesShort Note Biology Form 5-Chapter 3 Coordination and Responsesalamah_sabri75% (4)
- Jagaroo & Santangelo - Neurophenotypes Advancing Psychiatry and Neuropsychology 2016Document305 pagesJagaroo & Santangelo - Neurophenotypes Advancing Psychiatry and Neuropsychology 2016Jaime Fernández-Aguirrebengoa100% (1)
- MHE RDG Wonders LVRDR G3 ELL U4W3 18Document24 pagesMHE RDG Wonders LVRDR G3 ELL U4W3 18Kenneth N AngelineNo ratings yet
- Trabalho em Saúde e A Implantação Do Acolhimento Na Atenção Primária À Saúde - Afeto, Empatia Ou AlteridadeDocument13 pagesTrabalho em Saúde e A Implantação Do Acolhimento Na Atenção Primária À Saúde - Afeto, Empatia Ou AlteridadeAmérico PastorNo ratings yet
- Update On Perioperative Fluid Therapy DR Y Narendra, SPBDocument36 pagesUpdate On Perioperative Fluid Therapy DR Y Narendra, SPBnafisyarifahNo ratings yet
- Taxonomy of Haematococcus PluvialisDocument2 pagesTaxonomy of Haematococcus PluvialisKomathi BalasupramaniamNo ratings yet
- Personal Development Lesson 2Document54 pagesPersonal Development Lesson 2Ysay FranciscoNo ratings yet
- Penilaian Sumatif Satuan Pendidikan (PSSP) Mata Pelajaran Bahasa Inggris Ta. 20222023Document1 pagePenilaian Sumatif Satuan Pendidikan (PSSP) Mata Pelajaran Bahasa Inggris Ta. 20222023Malfa FaradisNo ratings yet
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeRating: 2 out of 5 stars2/5 (1)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (42)
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (24)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (3)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 5 out of 5 stars5/5 (80)
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 4 out of 5 stars4/5 (5)
- Summary: The Myth of Normal: Trauma, Illness, and Healing in a Toxic Culture By Gabor Maté MD & Daniel Maté: Key Takeaways, Summary & AnalysisFrom EverandSummary: The Myth of Normal: Trauma, Illness, and Healing in a Toxic Culture By Gabor Maté MD & Daniel Maté: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (9)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- Sleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningFrom EverandSleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningRating: 4 out of 5 stars4/5 (3)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (266)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisFrom EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisRating: 3.5 out of 5 stars3.5/5 (2)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDFrom EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDRating: 5 out of 5 stars5/5 (1)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 3.5 out of 5 stars3.5/5 (3)
- Outlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisFrom EverandOutlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (1)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessFrom EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessRating: 4.5 out of 5 stars4.5/5 (328)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeFrom EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeRating: 4.5 out of 5 stars4.5/5 (253)
- Gut: the new and revised Sunday Times bestsellerFrom EverandGut: the new and revised Sunday Times bestsellerRating: 4 out of 5 stars4/5 (392)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (169)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- To Explain the World: The Discovery of Modern ScienceFrom EverandTo Explain the World: The Discovery of Modern ScienceRating: 3.5 out of 5 stars3.5/5 (51)
- An Autobiography of Trauma: A Healing JourneyFrom EverandAn Autobiography of Trauma: A Healing JourneyRating: 5 out of 5 stars5/5 (2)
- 12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosFrom Everand12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosRating: 4.5 out of 5 stars4.5/5 (207)