You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5819)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Alcohols Phenols and EthersDocument1 pageAlcohols Phenols and EthersNitisha GuptaNo ratings yet
- Energy Conversion and Management: Rabah GomriDocument9 pagesEnergy Conversion and Management: Rabah GomriStanley PrimoNo ratings yet
- 2nd Preliminary Examination 2023 - 2024 Grade 10 ScienceDocument2 pages2nd Preliminary Examination 2023 - 2024 Grade 10 ScienceJerome Tala-ocNo ratings yet
- Solution, Solubility & Gas LawsDocument5 pagesSolution, Solubility & Gas LawsSabbir HossainNo ratings yet
- HPMC CelluloseDocument32 pagesHPMC Cellulosemailtorubal2573No ratings yet
- NOTES-The Cyclohexane Ring FlipDocument5 pagesNOTES-The Cyclohexane Ring FlipAshwini Singh TomarNo ratings yet
- Tutorial Week 7 - QUESTION - DEC2017Document1 pageTutorial Week 7 - QUESTION - DEC2017Shazlen AmranNo ratings yet
- Kinetics Lab Explained - Iodination of Acetone - Online Homework Help - SchoolWorkHelper PDFDocument4 pagesKinetics Lab Explained - Iodination of Acetone - Online Homework Help - SchoolWorkHelper PDFRajesh Shahi0% (1)
- Lab 3: Solubility of Organic Compounds: ObjectivesDocument12 pagesLab 3: Solubility of Organic Compounds: ObjectivesJAN CAMILLE OLIVARESNo ratings yet
- Surface Failures & Different Types of WearDocument12 pagesSurface Failures & Different Types of WearsathvikNo ratings yet
- Chemistry Project On Amount of Acetic Acid in VinegarDocument23 pagesChemistry Project On Amount of Acetic Acid in VinegarPratik DhondeNo ratings yet
- Precipitation Reactions Chapter 14Document68 pagesPrecipitation Reactions Chapter 14delialbuNo ratings yet
- ReportDocument30 pagesReportapi-439738433No ratings yet
- UNIT 4 Metal FinishingDocument17 pagesUNIT 4 Metal FinishingVasudev GuptaNo ratings yet
- Worksheet Holiday Homework Class 10-1Document2 pagesWorksheet Holiday Homework Class 10-1Himanshu YadavNo ratings yet
- A Level Chemistry Core Practical 16 - AspirinDocument5 pagesA Level Chemistry Core Practical 16 - Aspirinelsiesaveena96No ratings yet
- R&ACDocument2 pagesR&ACsubramanian jNo ratings yet
- Lámina ParacetamolDocument2 pagesLámina ParacetamolAndrea Alvarado RoNo ratings yet
- Textbook Ebook Electrical Properties of Materials 10Th Edition Solymar All Chapter PDFDocument43 pagesTextbook Ebook Electrical Properties of Materials 10Th Edition Solymar All Chapter PDFjulia.barhorst499100% (11)
- Neet 2023 Question Paper f4Document51 pagesNeet 2023 Question Paper f4Nitin ChahalNo ratings yet
- Cooling Tower SeminarDocument67 pagesCooling Tower SeminarRupesh Desai100% (1)
- MSE8013 - Chapter03 - Atomic BindingDocument37 pagesMSE8013 - Chapter03 - Atomic BindingJie GanNo ratings yet
- Hydrochloric Acid AND Miscellaneous Inorganic ChemicalsDocument9 pagesHydrochloric Acid AND Miscellaneous Inorganic ChemicalsHoneylet Recaña TayactacNo ratings yet
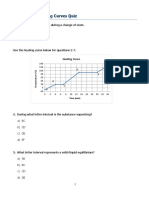
- Heating and Cooling Curves QuizDocument3 pagesHeating and Cooling Curves QuizAri ChristianNo ratings yet
- Disadvantages of Hard Water Boiler TroublesDocument6 pagesDisadvantages of Hard Water Boiler Troubleskhushboo goyalNo ratings yet
- Amadeus User GuideDocument54 pagesAmadeus User GuideIsmail OrakNo ratings yet
- Value Added Products From Gasification Activated Carbon: P. J. PaulDocument3 pagesValue Added Products From Gasification Activated Carbon: P. J. Paullpumsun1No ratings yet
- Inorganic and Analytical Chemistry CH-102 Engr. Muhammad Arslan HaiderDocument38 pagesInorganic and Analytical Chemistry CH-102 Engr. Muhammad Arslan Haiderinam ullahNo ratings yet
- Vanders Human Physiology The Mechanisms of Body Function 13th Edition Widmaier Test BankDocument36 pagesVanders Human Physiology The Mechanisms of Body Function 13th Edition Widmaier Test Bankmungoosemodus1qrzsk100% (18)
- CHROMATOGRAPHYDocument14 pagesCHROMATOGRAPHYBrian PaguiaNo ratings yet