You might also like
- Ansys Tutorial Forthe Torque Analysis of The Shaft Attached With Two DisksDocument13 pagesAnsys Tutorial Forthe Torque Analysis of The Shaft Attached With Two Disksvinvia100% (1)
- Problem Description: Problem 8: Analysis of A Shell CornerDocument14 pagesProblem Description: Problem 8: Analysis of A Shell Corneryoki_triwahyudiNo ratings yet
- Tutorial 28 Coal Mine StopeDocument18 pagesTutorial 28 Coal Mine Stoperongow titoNo ratings yet
- Tanner Tutorial CMOS NAND2Document11 pagesTanner Tutorial CMOS NAND2mdzakir_hussainNo ratings yet
- Introduction To Electrostatic FEA With BELADocument9 pagesIntroduction To Electrostatic FEA With BELAASOCIACION ATECUBONo ratings yet
- Ansys Vibes1Document19 pagesAnsys Vibes1api-3833671No ratings yet
- Lab2 Inverter S EditDocument17 pagesLab2 Inverter S EditumairazulkifliNo ratings yet
- Tutorial RubisDocument22 pagesTutorial RubisTeban CastroNo ratings yet
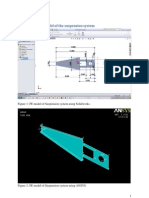
- Compressible Flow in A NozzleDocument6 pagesCompressible Flow in A NozzleAbhishek MeNo ratings yet
- KAPPA Rubis TutorialDocument22 pagesKAPPA Rubis TutorialSARTHAK BAPAT100% (1)
- Ansys Tutorial Forthe Torque Analysis of The Shaft Attached With Two DisksDocument13 pagesAnsys Tutorial Forthe Torque Analysis of The Shaft Attached With Two DisksPugazhenthi ThananjayanNo ratings yet
- TCAD Workshop Volume IDocument69 pagesTCAD Workshop Volume IVipan SharmaNo ratings yet
- Module XLC - Additional Information On Graphs in EXCEL Learning ObjectivesDocument13 pagesModule XLC - Additional Information On Graphs in EXCEL Learning ObjectivesM VetriselviNo ratings yet
- Analytical Modelling (Ansys)Document29 pagesAnalytical Modelling (Ansys)Izzah Yahya IINo ratings yet
- PSpice Tutorial PDFDocument24 pagesPSpice Tutorial PDFbhushanatkarmic100% (1)
- Turbulent Flow PDFDocument18 pagesTurbulent Flow PDFVinod Kumar PatelNo ratings yet
- B03 - Numex01Document29 pagesB03 - Numex01KARARNo ratings yet
- ANSYS Axial Bar TutorialDocument18 pagesANSYS Axial Bar TutorialappaduraiNo ratings yet
- Tutorial Week 3c - MECH3361 Workbench Guide PDFDocument12 pagesTutorial Week 3c - MECH3361 Workbench Guide PDFVijendraAgarNo ratings yet
- Cad Exp-6Document12 pagesCad Exp-6Harshal DodkeNo ratings yet
- Lesson08 3D FEM ClevisDocument10 pagesLesson08 3D FEM Clevisaiyubi2No ratings yet
- Introduction To ScicosLab-ScicosDocument33 pagesIntroduction To ScicosLab-ScicosJeziel JuárezNo ratings yet
- A Short Introduction To CADENCE Virtuoso Front To Back Design EnvironmentDocument25 pagesA Short Introduction To CADENCE Virtuoso Front To Back Design EnvironmentandroidNo ratings yet
- ANSYS Tutorial-Crack ProblemDocument8 pagesANSYS Tutorial-Crack ProblemMahdi100% (3)
- VMODFlex UsersManualm138-207Document70 pagesVMODFlex UsersManualm138-207Adrian Apaza LeomNo ratings yet
- Caelinux and Finite Element Analysis. Tutorial 1, Perforated CubeDocument14 pagesCaelinux and Finite Element Analysis. Tutorial 1, Perforated Cubewindsurferke007No ratings yet
- Guide To TannerEDA For VLSIDocument32 pagesGuide To TannerEDA For VLSINimit AroraNo ratings yet
- 2D Analysis of Cantilver Beam Subjected To Point LoadDocument14 pages2D Analysis of Cantilver Beam Subjected To Point LoadNEELIMANo ratings yet
- Workshop 09 B Post-ProcessingDocument42 pagesWorkshop 09 B Post-ProcessingIgnacio León IbarraNo ratings yet
- Lab 1 Cadence TutorialDocument36 pagesLab 1 Cadence TutorialN Nanda GaneshNo ratings yet
- Tutorial Everfe Ex01 02Document20 pagesTutorial Everfe Ex01 02rasec1989No ratings yet
- Tutorial 3 - Crack ProblemDocument8 pagesTutorial 3 - Crack ProblemImran2109No ratings yet
- Ansys Statics2 v61Document7 pagesAnsys Statics2 v61api-3833671No ratings yet
- FRAC W01 ThreePointDocument11 pagesFRAC W01 ThreePointavenashaNo ratings yet
- ADS TutorialDocument7 pagesADS TutorialNithesh Chakravarthi NekkantiNo ratings yet
- EE115C - Digital Electronic Circuits Tutorial 1.2: Getting Familiar With Technology, MOS IV CharacteristicsDocument16 pagesEE115C - Digital Electronic Circuits Tutorial 1.2: Getting Familiar With Technology, MOS IV CharacteristicsAhmed EdrisNo ratings yet
- Paschal's Intensive Course ReportDocument176 pagesPaschal's Intensive Course ReportSamuel charlesNo ratings yet
- Labs 1/2: Getting Started.: (3.0) - Version 5.0 Has Difficulties With Visualizing Orbitals in The Friezen Lab. The OlderDocument9 pagesLabs 1/2: Getting Started.: (3.0) - Version 5.0 Has Difficulties With Visualizing Orbitals in The Friezen Lab. The OlderSalah AlchimistNo ratings yet
- Cadence IntroductionDocument15 pagesCadence Introductiondragos_bondNo ratings yet
- RD-2100-Nonlinear AnalysisDocument11 pagesRD-2100-Nonlinear AnalysisShaheen S. RatnaniNo ratings yet
- ChemicalTankerTutorial ExpressMarine V1.2.1Document129 pagesChemicalTankerTutorial ExpressMarine V1.2.1Ante CrnicaNo ratings yet
- Icepak v12.0 Tut 03Document26 pagesIcepak v12.0 Tut 03Hassen HoussiNo ratings yet
- Pressure Block Study With Cosmosworks: File Open PV - 1 - 8.SldprtDocument19 pagesPressure Block Study With Cosmosworks: File Open PV - 1 - 8.SldprtbmyertekinNo ratings yet
- Lab #1 Introduction To LSDyna: Simple CantileverDocument22 pagesLab #1 Introduction To LSDyna: Simple CantileverMohammed A. MaherNo ratings yet
- Cad Exp-7Document12 pagesCad Exp-7Harshal DodkeNo ratings yet
- Splitting PartsDocument6 pagesSplitting PartsmikeboNo ratings yet
- Vensim StudyDocument8 pagesVensim StudydidikkrisNo ratings yet
- Excel VBA CodeDocument6 pagesExcel VBA CodemcmonizNo ratings yet
- BEEE1313 LAB 3 Presentation SlideDocument33 pagesBEEE1313 LAB 3 Presentation SlideEd ItrNo ratings yet
- PSCAD TutorialDocument42 pagesPSCAD Tutorialchenukap100% (1)
- Excel 2007 for Scientists and EngineersFrom EverandExcel 2007 for Scientists and EngineersRating: 4 out of 5 stars4/5 (2)
- Modeling and Simulation of Logistics Flows 3: Discrete and Continuous Flows in 2D/3DFrom EverandModeling and Simulation of Logistics Flows 3: Discrete and Continuous Flows in 2D/3DNo ratings yet
- Introduction to the simulation of power plants for EBSILON®Professional Version 15From EverandIntroduction to the simulation of power plants for EBSILON®Professional Version 15No ratings yet
- A Brief Introduction to MATLAB: Taken From the Book "MATLAB for Beginners: A Gentle Approach"From EverandA Brief Introduction to MATLAB: Taken From the Book "MATLAB for Beginners: A Gentle Approach"Rating: 2.5 out of 5 stars2.5/5 (2)
- PSS&C - PKundur - Cap06Document72 pagesPSS&C - PKundur - Cap06Francisco GuerraNo ratings yet
- EE276 Test 8Document3 pagesEE276 Test 8fred francisNo ratings yet
- 342 B.sc.b.ed. Mdsu PDF 4yrDocument135 pages342 B.sc.b.ed. Mdsu PDF 4yrDINESH SALVINo ratings yet
- Stainless Steel in Fire Final Summary Report March 08Document121 pagesStainless Steel in Fire Final Summary Report March 08twinpixtwinpixNo ratings yet
- Flame Photometer: Models PFP7 and PFP7/C Operating and Service ManualDocument45 pagesFlame Photometer: Models PFP7 and PFP7/C Operating and Service ManualSree Nivas ReddyNo ratings yet
- PCMDocument48 pagesPCMMushahary AnupNo ratings yet
- Lewis David On The Plurality of WorldsDocument279 pagesLewis David On The Plurality of Worldsdjoseph_1No ratings yet
- 1.constrution of Flying Quad Rotor With Video Surveillance SystemDocument45 pages1.constrution of Flying Quad Rotor With Video Surveillance SystemakhilNo ratings yet
- 10cordination CompDocument22 pages10cordination Compaleena'No ratings yet
- SIMS ManuscriptDocument6 pagesSIMS ManuscriptViraj EdirisingheNo ratings yet
- Evaluation of Deterioration and Restoration of The Limestone, White Monastery-Sohag, EgyptDocument17 pagesEvaluation of Deterioration and Restoration of The Limestone, White Monastery-Sohag, Egyptmarianascm26_9294298No ratings yet
- LV TrafoDocument38 pagesLV TrafoApik SubagyaNo ratings yet
- Science Chapter 12 Solar SystemDocument23 pagesScience Chapter 12 Solar Systemg-32347797No ratings yet
- Book - 2020 - M Tabatabaian - Tensor Analysis For Engineers PDFDocument197 pagesBook - 2020 - M Tabatabaian - Tensor Analysis For Engineers PDFVijay Kumar PolimeruNo ratings yet
- Olympus CX - 21 Manual de Usuario PDFDocument28 pagesOlympus CX - 21 Manual de Usuario PDFApocalipsis2072No ratings yet
- Locacion de Componentes ISXDocument9 pagesLocacion de Componentes ISXErick Lopez HdzNo ratings yet
- The Colonization of Tiamat V (Phoenix III, Daniel) PDFDocument44 pagesThe Colonization of Tiamat V (Phoenix III, Daniel) PDFKonstantina GlezakouNo ratings yet
- C26x Enm C57a PDFDocument1,284 pagesC26x Enm C57a PDFmn090078dNo ratings yet
- Chapter 17 - Essential Mathematical Methods Unit 1&2Document49 pagesChapter 17 - Essential Mathematical Methods Unit 1&2Kelley0% (1)
- Salsbury: Torque ConvertersDocument19 pagesSalsbury: Torque Convertersvt133primNo ratings yet
- Lecture 08 - ME 243 - Helical SpringsDocument14 pagesLecture 08 - ME 243 - Helical Springssalmanalamj5No ratings yet
- Is - 00191 - 2007 PDFDocument14 pagesIs - 00191 - 2007 PDFSangita GhaisasNo ratings yet
- Aim To Prepare A Sample of Cuprammonium Rayon Threads From Filter Paper Apparatus Required ADocument11 pagesAim To Prepare A Sample of Cuprammonium Rayon Threads From Filter Paper Apparatus Required ANitinAgnihotriNo ratings yet
- CBM Manual For PEDocument164 pagesCBM Manual For PEFaris Hazim100% (1)
- Hemp Thermographic ReportDocument20 pagesHemp Thermographic ReportEste BanNo ratings yet
- SAMPLE PAPER-AT-2324-C-XII-PASS-AT+PCM-Paper-2Document24 pagesSAMPLE PAPER-AT-2324-C-XII-PASS-AT+PCM-Paper-2Arijit DasNo ratings yet
- Reinforced Concrete Design TheoryDocument13 pagesReinforced Concrete Design Theorydragados7282150% (4)
- DCR Sr50eDocument77 pagesDCR Sr50epromatis0% (1)
- Chapter 2 Concrete ManualDocument10 pagesChapter 2 Concrete ManualnguyenkhachiepvnNo ratings yet
- Elec Motors Generators Design GuideDocument18 pagesElec Motors Generators Design GuidemiasatoNo ratings yet