You might also like
- Chapter 1 - Cellular Injury, Cell Adaptation & Cell Death (Robbins and Cotran Pathologic Basis of Disease)Document3 pagesChapter 1 - Cellular Injury, Cell Adaptation & Cell Death (Robbins and Cotran Pathologic Basis of Disease)Ernie G. Bautista II, RN, MD91% (34)
- Guillain Barre Syndrome-PPT FinalDocument39 pagesGuillain Barre Syndrome-PPT Finalmeghana100% (1)
- Hormones and BehaviourDocument367 pagesHormones and BehaviourRojo100% (3)
- Ministry of Education and Scientific Research) ) Al-Muthanna University College of Medicine) )Document10 pagesMinistry of Education and Scientific Research) ) Al-Muthanna University College of Medicine) )Hussein H. MahmoodNo ratings yet
- Podocyte InjuryDocument17 pagesPodocyte Injurygalihrahman51No ratings yet
- Minimal Change DiseaseDocument72 pagesMinimal Change DiseasekamalNo ratings yet
- Path Physiology of Sepsis. SIRS & MOFDocument7 pagesPath Physiology of Sepsis. SIRS & MOF18zyyNo ratings yet
- Genetic DiseasesDocument108 pagesGenetic DiseasesRizki Widya NurNo ratings yet
- Genetic Basis of Steroid Resistant Nephrotic Syndrome: Review ArticleDocument7 pagesGenetic Basis of Steroid Resistant Nephrotic Syndrome: Review ArticleIgnacio TabuadaNo ratings yet
- Chronic Intestinal PseudoDocument10 pagesChronic Intestinal PseudoErgueta SergioNo ratings yet
- Pediatric Nephrotic Nephritic SyndromesDocument18 pagesPediatric Nephrotic Nephritic SyndromesBryan AtasNo ratings yet
- Jurnal ReadingDocument34 pagesJurnal ReadingFerdian HayafiNo ratings yet
- Glomerular Podocyte Dysfunction in Inherited Renal Tubular DiseaseDocument7 pagesGlomerular Podocyte Dysfunction in Inherited Renal Tubular DiseaseAkhemi NakamuraNo ratings yet
- Podocyte Disorders Core Curriculum 2011 - American Journal of Kidney DiseasesDocument1 pagePodocyte Disorders Core Curriculum 2011 - American Journal of Kidney DiseasesYunitaNo ratings yet
- Nephrotic Syndrome HandoutDocument10 pagesNephrotic Syndrome HandoutRizna SaidNo ratings yet
- Podocyte Biology and The EmergingDocument8 pagesPodocyte Biology and The Emerging_siegfried_No ratings yet
- Current Status of Gene Therapy Research in PolyglutamineDocument32 pagesCurrent Status of Gene Therapy Research in PolyglutamineTanzeela noureenNo ratings yet
- Multiple Organ Dysfunction SyndromeDocument21 pagesMultiple Organ Dysfunction SyndromeRo-Anne LozadaNo ratings yet
- Diseases of Urinary SystemDocument29 pagesDiseases of Urinary SystemHassan.shehri100% (9)
- Cancer BiologyDocument32 pagesCancer BiologyEmre AktaşNo ratings yet
- Biomol SN InvivoDocument11 pagesBiomol SN Invivogalihrahman51No ratings yet
- 131102.2 20200108132649 CoveredDocument18 pages131102.2 20200108132649 Covered6pb4rwpk45No ratings yet
- Primer: PodocytopathiesDocument24 pagesPrimer: PodocytopathiesMohamad IrwanNo ratings yet
- 18 NephroticsyndromeDocument7 pages18 NephroticsyndromeKim's FamilyNo ratings yet
- Robbins File For AnkiDocument13 pagesRobbins File For Ankiאיתי עוזרNo ratings yet
- Genetic Toxic and Acquired Metabolic DiseasesDocument37 pagesGenetic Toxic and Acquired Metabolic DiseasesQuenee Joyce CabelloNo ratings yet
- Main (2) Manif+terapii MedDocument13 pagesMain (2) Manif+terapii MedCosmin ChelariuNo ratings yet
- NonMalignant Leukocyte DisordersDocument43 pagesNonMalignant Leukocyte DisordersKei Ef SiNo ratings yet
- The Pathogenesis of Liver Cirrhosis / Fibrosis: Dr. Indranil BhattacharyaDocument46 pagesThe Pathogenesis of Liver Cirrhosis / Fibrosis: Dr. Indranil BhattacharyaDr. Indranil BhattacharayaNo ratings yet
- Nephrotic Syndrome in Children: January 2013Document7 pagesNephrotic Syndrome in Children: January 2013molenNo ratings yet
- CADAE1405CDocument12 pagesCADAE1405CMuhammad RiduanNo ratings yet
- Subcellular Organelles DiseasesDocument38 pagesSubcellular Organelles DiseasesAttiba RizwanNo ratings yet
- Histopathlogy General Concept Part 1Document58 pagesHistopathlogy General Concept Part 1Kimberly AnnNo ratings yet
- Introduction To Pathology Cmu 605 (Pathology Cmu 605) 2021 Enugu (Autosaved)Document355 pagesIntroduction To Pathology Cmu 605 (Pathology Cmu 605) 2021 Enugu (Autosaved)Ogbuefi PascalNo ratings yet
- CKD - Signaling Pathways of Chronic Kidney Diseases, Implications For TherapeuticsDocument27 pagesCKD - Signaling Pathways of Chronic Kidney Diseases, Implications For TherapeuticsCher IshNo ratings yet
- Disorders of Granulocytes and MonocytesDocument45 pagesDisorders of Granulocytes and Monocytesswathi bsNo ratings yet
- Hereditary Hemolytic Anaemia: Membrane and Enzymopathies DefectsDocument47 pagesHereditary Hemolytic Anaemia: Membrane and Enzymopathies DefectsaymenNo ratings yet
- Genetics Lec NotesDocument4 pagesGenetics Lec Notesellaph916No ratings yet
- Genetic and Pediatric DiseasesDocument88 pagesGenetic and Pediatric DiseasesJaneNo ratings yet
- Alternative Mechanisms of Signal Transduction: The Cellular and Molecular Basis of DevelopmentDocument32 pagesAlternative Mechanisms of Signal Transduction: The Cellular and Molecular Basis of DevelopmentDon MARSHALNo ratings yet
- Neurodegenerative Diseases Multifactorial ConformationalDocument9 pagesNeurodegenerative Diseases Multifactorial ConformationalLala FemNo ratings yet
- Nephrotic Syndrome PirDocument15 pagesNephrotic Syndrome PirNosirova ManijaNo ratings yet
- Neurodegener RalucaDocument88 pagesNeurodegener RalucaAlexandra AnaellyNo ratings yet
- Gefs Etiologia Agatti Conh 12Document8 pagesGefs Etiologia Agatti Conh 12Leoncio SerranoNo ratings yet
- Cell InjuryDocument39 pagesCell InjuryZainul Iman Abdul MalekNo ratings yet
- Pathology Biochemical and Molecular Basis of Some Mendelian DisordersDocument5 pagesPathology Biochemical and Molecular Basis of Some Mendelian DisordersAmna Baig0% (1)
- Fibronectin KadarDocument5 pagesFibronectin KadarVinnie Juliana YonatanNo ratings yet
- Recent Advances in Medicine and BiochemistryDocument28 pagesRecent Advances in Medicine and BiochemistryAaron JoseNo ratings yet
- Nefrotik SyndDocument13 pagesNefrotik SyndJorianditha RamadhanNo ratings yet
- Chapter One Introduction To PathologyDocument26 pagesChapter One Introduction To Pathologyrambabs369No ratings yet
- Chapter One Introduction To Pathology Chapter One Introduction To PathologyDocument26 pagesChapter One Introduction To Pathology Chapter One Introduction To PathologySivanarayana JayavaramNo ratings yet
- Enfermedad Renal y Desordenes HematologicasDocument11 pagesEnfermedad Renal y Desordenes HematologicasIris GzlzNo ratings yet
- 6.cell Membrane DiseasesDocument40 pages6.cell Membrane DiseasesStanley ChikoveNo ratings yet
- Cyclosporin in Idiopathic Glomerular Disease AssocDocument20 pagesCyclosporin in Idiopathic Glomerular Disease AssocsamudraandiNo ratings yet
- Dystrophy Apoptosis NecrosisDocument41 pagesDystrophy Apoptosis NecrosisAbuFreihNo ratings yet
- Hereditary Spherocytosis - UpToDateDocument76 pagesHereditary Spherocytosis - UpToDateHuỳnh Thị Khả DuyNo ratings yet
- Cell PathophysiologyDocument11 pagesCell PathophysiologyDURJOYNo ratings yet
- Glomerular DiseasesDocument92 pagesGlomerular Diseasesfrankozed1No ratings yet
- Membrane Lipid Signaling in Aging and Age-Related DiseaseFrom EverandMembrane Lipid Signaling in Aging and Age-Related DiseaseNo ratings yet
- Epigenetics of Aging and Longevity: Translational Epigenetics vol 4From EverandEpigenetics of Aging and Longevity: Translational Epigenetics vol 4Alexey MoskalevNo ratings yet
- Genetic Diagnosis of Endocrine DisordersFrom EverandGenetic Diagnosis of Endocrine DisordersRoy E. WeissNo ratings yet
- Biomechanics of NervesDocument65 pagesBiomechanics of NervesZinneRah RahManNo ratings yet
- Neurobiology - Option - IBDocument49 pagesNeurobiology - Option - IBJarek OleszczynskiNo ratings yet
- Enriquez Physio Ex3 - Act 7Document6 pagesEnriquez Physio Ex3 - Act 7Vergel Jigs EnriquezNo ratings yet
- Medical SurgicalDocument119 pagesMedical SurgicalNursyNurseNo ratings yet
- Osmosis High-Yield Physiology - Medicalstudyzone - Com (Sürüklenen)Document87 pagesOsmosis High-Yield Physiology - Medicalstudyzone - Com (Sürüklenen)Dilay MercimekNo ratings yet
- DONE BIO MOD Nervous System PDFDocument9 pagesDONE BIO MOD Nervous System PDFCristie SumbillaNo ratings yet
- Multiple SclerosisDocument4 pagesMultiple SclerosisnasibdinNo ratings yet
- Local Anesthesia I LectureDocument44 pagesLocal Anesthesia I LectureMavisNo ratings yet
- 2015 Bookmatter FundamentalsOfNeurologicDiseasDocument18 pages2015 Bookmatter FundamentalsOfNeurologicDiseasSabrin BadarneNo ratings yet
- Neurons, Synapses, and Signaling: Lecture Presentations by Nicole Tunbridge and Kathleen FitzpatrickDocument102 pagesNeurons, Synapses, and Signaling: Lecture Presentations by Nicole Tunbridge and Kathleen FitzpatrickSallyNo ratings yet
- Nervous System - HistologyDocument8 pagesNervous System - HistologyambercrisologoNo ratings yet
- 2022 Specimen Paper 4 Mark SchemeDocument20 pages2022 Specimen Paper 4 Mark Schemeadel hanyNo ratings yet
- Ana NervDocument5 pagesAna NervBrent ValdespinaNo ratings yet
- 28 - White Matter DiseasesDocument92 pages28 - White Matter DiseasesDARWING PADILLANo ratings yet
- OCR F214 MS Biology - Communication, Homeostasis and Energy January 2011 MARK SCHEMEDocument21 pagesOCR F214 MS Biology - Communication, Homeostasis and Energy January 2011 MARK SCHEMEDNo ratings yet
- Nervous System ANAPHY NotesDocument10 pagesNervous System ANAPHY NotesAlloiza CaguiclaNo ratings yet
- Practice Test 2 PDFDocument23 pagesPractice Test 2 PDFPremanshu SinghNo ratings yet
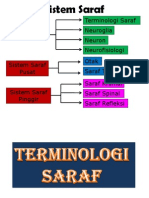
- Tisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafDocument141 pagesTisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafRainne LeeNo ratings yet
- Poly NeuropathyDocument42 pagesPoly NeuropathyNurmusofa Wibowo100% (1)
- Nerve ImpulseDocument8 pagesNerve ImpulseAsadNo ratings yet
- Final Examination: Organ Is TheDocument12 pagesFinal Examination: Organ Is TheAri BrownstoneNo ratings yet
- Double-Crush Syndrome - A Critical AnalysisDocument9 pagesDouble-Crush Syndrome - A Critical AnalysisCelina Tam Ching LamNo ratings yet
- NCM 116 - Neuro (MODULE 1)Document10 pagesNCM 116 - Neuro (MODULE 1)Meryville Jacildo100% (1)
- Report of Nerves TissueDocument20 pagesReport of Nerves Tissuerheny_greenNo ratings yet
- Happ Chapter 8 TransesDocument13 pagesHapp Chapter 8 TransesFrencess Kaye SimonNo ratings yet
- Cognition 6th Edition Ashcraft Test BankDocument35 pagesCognition 6th Edition Ashcraft Test Bankmaygluish5leo38100% (20)
- Nervous CoordinationDocument45 pagesNervous CoordinationTajXNo ratings yet
- Sistem Saraf PeriferDocument5 pagesSistem Saraf PeriferFitri SukmawatiNo ratings yet