You might also like
- Protein Purification and DeterminationDocument30 pagesProtein Purification and DeterminationNurl Aina100% (1)
- The Electricity Wiring RegulationDocument239 pagesThe Electricity Wiring Regulationgodson50% (2)
- Script of Cca Eim EditedDocument4 pagesScript of Cca Eim EditedMhen Maugan100% (1)
- Electrophoresis & Gel TechniquesDocument7 pagesElectrophoresis & Gel TechniquesnavedNo ratings yet
- DNA Repair: Chapter FourDocument7 pagesDNA Repair: Chapter FourMatin Ahmad KhanNo ratings yet
- Rate of ChangeDocument22 pagesRate of ChangeTrisha MariehNo ratings yet
- S.Y.Bsc Semester Iii Botany Paper IiDocument53 pagesS.Y.Bsc Semester Iii Botany Paper IiĐỗ Quang BìnhNo ratings yet
- Electrophoretic Mobility Shift Assay - Wikipedia, The Free EncyclopediaDocument3 pagesElectrophoretic Mobility Shift Assay - Wikipedia, The Free EncyclopediaShailendra YadavNo ratings yet
- 1.1 Proteins - Motifs Structural and Functional Domains Protein FamiliesDocument32 pages1.1 Proteins - Motifs Structural and Functional Domains Protein FamilieskinjalkaNo ratings yet
- SRT95 Off-Highway Truck Rear Axle Assembly: Structure and Installation Part 2 Parts Presentation Part 3 MaintenanceDocument29 pagesSRT95 Off-Highway Truck Rear Axle Assembly: Structure and Installation Part 2 Parts Presentation Part 3 MaintenanceoktopusNo ratings yet
- Dna SupercoilingDocument16 pagesDna SupercoilingrainabtNo ratings yet
- Agarose Gel Electrophoresis of DNA PDFDocument5 pagesAgarose Gel Electrophoresis of DNA PDFBiologistAhmedNo ratings yet
- Dna RepairDocument20 pagesDna RepairEaron Van JaboliNo ratings yet
- Study Notes: The GC ColumnDocument16 pagesStudy Notes: The GC ColumnLaxmi Kant PrasadNo ratings yet
- Unit 4 Chromatography-IIDocument18 pagesUnit 4 Chromatography-IIAli SheikhNo ratings yet
- Lab 8 - Transcription-Translation-ONLINE VERSION - 2021Document11 pagesLab 8 - Transcription-Translation-ONLINE VERSION - 2021thesoccerprince.10No ratings yet
- Prepared By: Verna Jean M. Magdayao 3/Bsbiology/ADocument56 pagesPrepared By: Verna Jean M. Magdayao 3/Bsbiology/AKathleya PeñaNo ratings yet
- Ejc-M13 - FN953205Document203 pagesEjc-M13 - FN953205JoséNo ratings yet
- Western Blotting MicrsoftDocument45 pagesWestern Blotting MicrsoftSoNu de Bond100% (2)
- Principles of Gel ElectrophoresisDocument6 pagesPrinciples of Gel ElectrophoresisCarmen Lopez100% (1)
- Calvin Cycle (Dark Reactions / Light Independent Reaction)Document47 pagesCalvin Cycle (Dark Reactions / Light Independent Reaction)L'ya Lieslotte100% (2)
- Spectrophotometry of DnaDocument7 pagesSpectrophotometry of DnaMel June FishNo ratings yet
- Electrophoresis Lecture 1Document38 pagesElectrophoresis Lecture 1Akor Pius udochukwu100% (1)
- Electrophoreti C Methods: Igaa SeptiariDocument22 pagesElectrophoreti C Methods: Igaa SeptiariGung Ari100% (1)
- Directed Mutagenesis and Protein EngineeringDocument52 pagesDirected Mutagenesis and Protein Engineeringslowdragon2003No ratings yet
- Astm D 2783 - 03Document9 pagesAstm D 2783 - 03Sérgio Luiz RodriguesNo ratings yet
- DNA Separation Using Agarose Gel ElectrophoresisDocument6 pagesDNA Separation Using Agarose Gel ElectrophoresisAnura BandaraNo ratings yet
- Agarose Gel ElectrophoresisDocument11 pagesAgarose Gel ElectrophoresisAbrar 111No ratings yet
- 05Document43 pages05Sania ZahoorNo ratings yet
- ElectrophoresisDocument45 pagesElectrophoresisPagla HowaNo ratings yet
- Comet AssayDocument15 pagesComet AssayarunsaintNo ratings yet
- Western BlottingDocument13 pagesWestern BlottingAshfaq Fazal100% (1)
- Phase Contrast MicrosDocument7 pagesPhase Contrast MicrosRahul PalsNo ratings yet
- What Is PyrosequencingDocument6 pagesWhat Is PyrosequencingAshish SachanNo ratings yet
- Immobilized Enzyme Kinetics Techniques and ApplicationsDocument92 pagesImmobilized Enzyme Kinetics Techniques and ApplicationsVNo ratings yet
- Gel FiltrationDocument123 pagesGel FiltrationZulfikri Asmardi RaufNo ratings yet
- Capillary Electrophoresis Vanessa CHEM6200Document32 pagesCapillary Electrophoresis Vanessa CHEM6200muldawatiNo ratings yet
- Dna StructureDocument23 pagesDna StructureAnonymous mHS76aNo ratings yet
- Protein PurificationDocument7 pagesProtein PurificationArchana BorahNo ratings yet
- Eukaryotic Expression SystemsDocument26 pagesEukaryotic Expression SystemsDeana NamirembeNo ratings yet
- BCH 314 Tutorial 1 SolutionsDocument9 pagesBCH 314 Tutorial 1 SolutionsvictorNo ratings yet
- Question 1 (37 Marks) : Biochemistry 3 BCH 314Document4 pagesQuestion 1 (37 Marks) : Biochemistry 3 BCH 314victorNo ratings yet
- 2016 BCH223 PracticalsDocument7 pages2016 BCH223 PracticalsvictorNo ratings yet
- Kami Export - Worksheet 14 Central DogmaDocument2 pagesKami Export - Worksheet 14 Central DogmaLindsey TamlinNo ratings yet
- Neurotransmitters - PPT 4Document39 pagesNeurotransmitters - PPT 4rikasusantiii100% (1)
- Restriction EnzymesDocument15 pagesRestriction EnzymesCharisma MeromiNo ratings yet
- DNA Quality-Spectrophotometry and ElectrophoresisDocument5 pagesDNA Quality-Spectrophotometry and Electrophoresislovina candra kirana100% (1)
- Gene PredictionDocument5 pagesGene PredictionNickson OnchokaNo ratings yet
- Gel filtration chromatography separates molecules based on sizeDocument16 pagesGel filtration chromatography separates molecules based on sizeGhassanM.Ahmed GhassanM.Ahmed100% (1)
- Restriction Enzyme DigestionDocument5 pagesRestriction Enzyme DigestionAqsa ImtiazNo ratings yet
- Plasmid DNA Quantification of DNADocument3 pagesPlasmid DNA Quantification of DNAHuishin LeeNo ratings yet
- Submitted by Aiswarya V 1St MSC Zoology Roll Number 3301Document32 pagesSubmitted by Aiswarya V 1St MSC Zoology Roll Number 3301PES PEOPLENo ratings yet
- Ultracentrifugation: June 2010Document8 pagesUltracentrifugation: June 2010Nayak Alok RanjanNo ratings yet
- Chemical and Physical Properties of Nucleic AcidsDocument6 pagesChemical and Physical Properties of Nucleic AcidsSherlock Wesley ConanNo ratings yet
- DNA Repair: Vipin ShankarDocument32 pagesDNA Repair: Vipin ShankarMaximilian Magulye100% (1)
- Types of Cell CultureDocument5 pagesTypes of Cell CultureSarah PavuNo ratings yet
- L 10 Post Transcriptional ModificationDocument33 pagesL 10 Post Transcriptional ModificationsNo ratings yet
- Ion ChromatographyDocument2 pagesIon ChromatographyalexpharmNo ratings yet
- Protein TargettingDocument11 pagesProtein TargettingPMIB Matrikulasi FKUI 2018/2019No ratings yet
- Unit 3 The Three Dimensional Structure of ProteinsDocument20 pagesUnit 3 The Three Dimensional Structure of ProteinsPatricia OrtizNo ratings yet
- Biomaterials Exam 2 HW Answer Key PDFDocument24 pagesBiomaterials Exam 2 HW Answer Key PDFChelle VillasisNo ratings yet
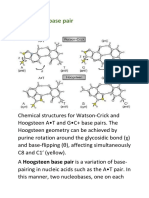
- Hoogsteen Base Pair PDFDocument7 pagesHoogsteen Base Pair PDFNikitaNo ratings yet
- Alkaline Phosphatase and LigasesDocument18 pagesAlkaline Phosphatase and LigasessathyaNo ratings yet
- Mechanism of Enzyme ActionDocument2 pagesMechanism of Enzyme ActionBlazy InhumangNo ratings yet
- Estimation of DNADocument1 pageEstimation of DNATjcbt BiosciencesNo ratings yet
- Company Electronics Appliances Semiconductors South Korea's: SamsungDocument61 pagesCompany Electronics Appliances Semiconductors South Korea's: Samsunghaseeb ahmedNo ratings yet
- Weap - ModflowDocument20 pagesWeap - Modflowguive3No ratings yet
- Food, Family, and FriendshipsDocument256 pagesFood, Family, and FriendshipsBianca PradoNo ratings yet
- Burkert General Catalogue Rev2Document44 pagesBurkert General Catalogue Rev2cuongNo ratings yet
- Quiz - Limits and ContinuityDocument3 pagesQuiz - Limits and ContinuityAdamNo ratings yet
- 水印AMSCO® Advanced Placement® European History, 水印2nd Edition (Perfection Learning Authors)Document764 pages水印AMSCO® Advanced Placement® European History, 水印2nd Edition (Perfection Learning Authors)Cherry panNo ratings yet
- Diagnosing and Managing Acute and Chronic SinusitisDocument14 pagesDiagnosing and Managing Acute and Chronic SinusitisAnonymous y3TIOwX8100% (1)
- PIRA - 2022 - ESTIMATED FMV As of October 2022Document48 pagesPIRA - 2022 - ESTIMATED FMV As of October 2022Aggy ReynadoNo ratings yet
- Delays in Endoscope Reprocessing and The Biofilms WithinDocument12 pagesDelays in Endoscope Reprocessing and The Biofilms WithinHAITHM MURSHEDNo ratings yet
- Literature Review On Waste Management in NigeriaDocument9 pagesLiterature Review On Waste Management in NigeriajzneaqwgfNo ratings yet
- Analog and Digital Communication Systems: The Transmission of Information Is Called CommunicationDocument71 pagesAnalog and Digital Communication Systems: The Transmission of Information Is Called Communication20-403 TejashwiniNo ratings yet
- Cash and Cash EquivalentsDocument2 pagesCash and Cash EquivalentsMary Jullianne Caile SalcedoNo ratings yet
- Business PolicyDocument17 pagesBusiness PolicyManish SinghNo ratings yet
- Pradeep Kumaresan Resume - Software Testing ExperienceDocument3 pagesPradeep Kumaresan Resume - Software Testing ExperienceSANTANo ratings yet
- Akuntansi Keuangan Lanjutan - Akuntansi Penggabungan UsahaDocument67 pagesAkuntansi Keuangan Lanjutan - Akuntansi Penggabungan UsahachendyNo ratings yet
- K - LP - Week 24 - Journeys Unit 3 Lesson 14Document8 pagesK - LP - Week 24 - Journeys Unit 3 Lesson 14englishwithmslilyNo ratings yet
- Water TreatmentDocument6 pagesWater TreatmentSantiago LarrazNo ratings yet
- Uconnect User GuideDocument113 pagesUconnect User GuidetamilarasansrtNo ratings yet
- KPW FSO Yetagun Presentation 250713Document36 pagesKPW FSO Yetagun Presentation 250713muhamadrafie1975No ratings yet
- Chemists 12-2023Document7 pagesChemists 12-2023PRC BaguioNo ratings yet
- Chapter 5 Perfect CompetitionDocument20 pagesChapter 5 Perfect Competition刘文雨杰No ratings yet
- Qualitative Research On Vocabulary and Spelling Skills of A Student Chapters I IIIDocument23 pagesQualitative Research On Vocabulary and Spelling Skills of A Student Chapters I IIIarniza blazoNo ratings yet
- Repport Btech FinalDocument49 pagesRepport Btech FinalSuzelle NGOUNOU MAGANo ratings yet
- UFD/MMC/SD Controller Flash Support Limitation and Interconnection NoteDocument5 pagesUFD/MMC/SD Controller Flash Support Limitation and Interconnection Noteمہرؤآنہ آبہرآهہيہمہNo ratings yet