You might also like
- Python for Chemistry: An introduction to Python algorithms, Simulations, and Programing for Chemistry (English Edition)From EverandPython for Chemistry: An introduction to Python algorithms, Simulations, and Programing for Chemistry (English Edition)Rating: 5 out of 5 stars5/5 (1)
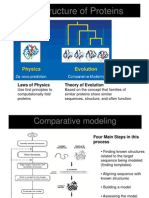
- Homology ModellingDocument21 pagesHomology ModellingJerome FrancisNo ratings yet
- Tertiary Structure Prediction Methods: Any Given Protein SequenceDocument29 pagesTertiary Structure Prediction Methods: Any Given Protein Sequencesurrender003No ratings yet
- Pre-Assessment QuestionsDocument18 pagesPre-Assessment Questionsk.sachinNo ratings yet
- Homology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersDocument16 pagesHomology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersPallavi VermaNo ratings yet
- Protein Structure Prediction TechniquesDocument30 pagesProtein Structure Prediction TechniquesJhanvi SNo ratings yet
- 3D Structure PredictionDocument33 pages3D Structure PredictionAyesha KhanNo ratings yet
- Homology Modeling Technique for Protein Structure PredictionDocument19 pagesHomology Modeling Technique for Protein Structure PredictionJainendra JainNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Homology Modelling: Structure Prediction Using TemplatesDocument19 pagesHomology Modelling: Structure Prediction Using TemplatesManoharNo ratings yet
- HomolgyDocument21 pagesHomolgySatish SahuNo ratings yet
- Protein Modelling MethodsDocument19 pagesProtein Modelling MethodsrednriNo ratings yet
- 3D Structure of Proteins Prediction MethodsDocument9 pages3D Structure of Proteins Prediction MethodsladykikyoyunaNo ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- Homologymodelingmodeller 181002182751Document19 pagesHomologymodelingmodeller 181002182751Jhanvi SNo ratings yet
- Homology ModellingDocument29 pagesHomology ModellingRISHITA CHAUHANNo ratings yet
- Homologymodeling 150123025144 Conversion Gate01Document27 pagesHomologymodeling 150123025144 Conversion Gate01Jhanvi SNo ratings yet
- Methods For Accurate Homology Modeling by Global OptimizationDocument17 pagesMethods For Accurate Homology Modeling by Global OptimizationUmmuNo ratings yet
- Re Comb 2003Document16 pagesRe Comb 2003mustafaNo ratings yet
- Protein ModelingDocument17 pagesProtein ModelingManeeshAgrawalNo ratings yet
- Protein Tertiaty Structure PredictionDocument12 pagesProtein Tertiaty Structure PredictionRoshan Mammen ChakkungalNo ratings yet
- Protein ModellingDocument15 pagesProtein ModellingNandini KotharkarNo ratings yet
- Roy Et Al. - 2011 - A Protocol For Computer-Based Protein Structure AnDocument10 pagesRoy Et Al. - 2011 - A Protocol For Computer-Based Protein Structure Anadiav PakNo ratings yet
- Insilico DockingDocument6 pagesInsilico DockingRajesh GuruNo ratings yet
- Prediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsDocument11 pagesPrediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsLata DeshmukhNo ratings yet
- Bif501 AssDocument3 pagesBif501 Asssana 568aNo ratings yet
- Homology Modelling: A 5-Step ProcessDocument30 pagesHomology Modelling: A 5-Step ProcesskamleshNo ratings yet
- SHARPEN Software for Protein Structure Prediction and DesignDocument10 pagesSHARPEN Software for Protein Structure Prediction and DesignSuchismita PatraNo ratings yet
- Modelling protein quaternary structure using Swiss-ModelDocument11 pagesModelling protein quaternary structure using Swiss-Model19BTBT037 S.VishaliNo ratings yet
- Identification of Modelling TemplatesDocument7 pagesIdentification of Modelling TemplatesLokesh AdhikariNo ratings yet
- Proclust:: Improved Clustering of Protein Sequences With An Extended Graph-Based ApproachDocument58 pagesProclust:: Improved Clustering of Protein Sequences With An Extended Graph-Based ApproachJJ EBNo ratings yet
- Lesson 2Document17 pagesLesson 2Vamsi Krishna ChaitanyaNo ratings yet
- Abhilash-SWISS MODEL Seminar 2023Document25 pagesAbhilash-SWISS MODEL Seminar 2023abhilashjgNo ratings yet
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Sanchez CurrOpinStructBiol 1997Document9 pagesSanchez CurrOpinStructBiol 1997Phoenix RisingNo ratings yet
- Generative Modeling for Protein StructuresDocument12 pagesGenerative Modeling for Protein Structurescarlos ArozamenaNo ratings yet
- Ab Initio Protein Structure PredictionDocument25 pagesAb Initio Protein Structure PredictionVishnu AjithNo ratings yet
- Protein Modeling by Multiple Sequence Threading and Distance GeometryDocument5 pagesProtein Modeling by Multiple Sequence Threading and Distance GeometryLata DeshmukhNo ratings yet
- Lecture 101Document43 pagesLecture 101Jean ReneNo ratings yet
- Structural Biology: What Does 3D Tell Us?Document20 pagesStructural Biology: What Does 3D Tell Us?Prakash KumarNo ratings yet
- 791 Ak Lecture6Document50 pages791 Ak Lecture6srikalyani2k9No ratings yet
- Structural BioinfoDocument76 pagesStructural BioinfoMudit MisraNo ratings yet
- Simulation-Based Automatic Generation Signomial and Posynomial Performance Models Analog Integrated Circuit SizingDocument5 pagesSimulation-Based Automatic Generation Signomial and Posynomial Performance Models Analog Integrated Circuit Sizingsuchi87No ratings yet
- 17 - Chapter 9 PDFDocument5 pages17 - Chapter 9 PDFما هذا الهراءNo ratings yet
- Automated Design of The Surface Positions of Protein HelicesDocument9 pagesAutomated Design of The Surface Positions of Protein Helicesaxva1663No ratings yet
- Protein-Ligand Docking: Dr. Noel O'Boyle University College Cork N.oboyle@ucc - IeDocument39 pagesProtein-Ligand Docking: Dr. Noel O'Boyle University College Cork N.oboyle@ucc - IeRustEdNo ratings yet
- Improved Particle Swarm Optimization For The Determination of Chaboche Model Parameters of The Elastoplastic Behavior Railway SteelDocument10 pagesImproved Particle Swarm Optimization For The Determination of Chaboche Model Parameters of The Elastoplastic Behavior Railway SteelInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- (Sumber: Radiation Oncology Physics: A Handbook For Teachers and Students) /podgorsakDocument4 pages(Sumber: Radiation Oncology Physics: A Handbook For Teachers and Students) /podgorsakYoza FendrianiNo ratings yet
- Molecular Modelling & Drug DesignDocument52 pagesMolecular Modelling & Drug DesignArthe RajarajanNo ratings yet
- 10 21105 Joss 05057Document5 pages10 21105 Joss 05057Arpanet ArpanetNo ratings yet
- Eswar MethodsMolBiol 2008Document25 pagesEswar MethodsMolBiol 2008huouinkyoumaNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- International Journal of Research and Development in Pharmacy and Life SciencesDocument9 pagesInternational Journal of Research and Development in Pharmacy and Life SciencesAkash NarayananNo ratings yet
- Protein Structure Prediction ThesisDocument8 pagesProtein Structure Prediction ThesisWriteMyThesisPaperManchester100% (2)
- Proteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingDocument10 pagesProteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingLavanya Priya SathyanNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- In Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeDocument8 pagesIn Silico Attributes of Cold Resistance Protein 1 (Crp1) F Rom Brassica OleraceaeresearchinventyNo ratings yet
- Docking & DynamicsDocument51 pagesDocking & DynamicsIqbal RadetyoNo ratings yet
- Multiple Sequence Alignment Part 1Document64 pagesMultiple Sequence Alignment Part 1letsvanshNo ratings yet
- Annurev Pharmtox 010814 124844Document28 pagesAnnurev Pharmtox 010814 124844Basab GhoshNo ratings yet
- Ultrasonic Flow Meter: Working PrincipleDocument1 pageUltrasonic Flow Meter: Working PrincipleBasab GhoshNo ratings yet
- Turbine Flow Meter: Working PrincipleDocument1 pageTurbine Flow Meter: Working PrincipleBasab GhoshNo ratings yet
- Site Directed MutagenesisDocument4 pagesSite Directed MutagenesisBasab GhoshNo ratings yet
- BiophysicsandMolecularBiology PDFDocument101 pagesBiophysicsandMolecularBiology PDFRisikrrishna DasNo ratings yet
- White Collar CrimesDocument201 pagesWhite Collar CrimesBasab GhoshNo ratings yet
- Alkyd Resin Soucek2015Document83 pagesAlkyd Resin Soucek2015Anchit GhoshNo ratings yet
- CompReg 25AUGUST2020Document5,032 pagesCompReg 25AUGUST2020starNo ratings yet
- Immuno Oncology Elisa Brochure PDFDocument8 pagesImmuno Oncology Elisa Brochure PDFSofia TheodorouNo ratings yet
- Test Bank For Molecular Diagnostics 3rd Edition Lela BuckinghamDocument21 pagesTest Bank For Molecular Diagnostics 3rd Edition Lela BuckinghamMadge Oliva100% (32)
- Dna TechDocument6 pagesDna TechReymond Jude PagcoNo ratings yet
- Human Genetics and Cloning Project Class 12Document21 pagesHuman Genetics and Cloning Project Class 12Taran OjhaNo ratings yet
- work: Progress OF ToDocument7 pageswork: Progress OF ToUsman DarNo ratings yet
- RNA CatalysisDocument37 pagesRNA CatalysisPREETI MANGALNo ratings yet
- ProteinDocument23 pagesProteinsamantha garciaNo ratings yet
- What Makes Us HumanDocument7 pagesWhat Makes Us HumanGabriela FloareNo ratings yet
- Eileen Naga - Isolation of DnaDocument4 pagesEileen Naga - Isolation of DnaEileen NagaNo ratings yet
- Penawaran PT Multisera IndosaDocument1 pagePenawaran PT Multisera IndosaKontol LodonNo ratings yet
- The Fundamental Unit of Life Class 9 Important Questions Science Chapter 5Document45 pagesThe Fundamental Unit of Life Class 9 Important Questions Science Chapter 5Brijesh DeviNo ratings yet
- CBE Courses Tables 1 and 2Document4 pagesCBE Courses Tables 1 and 2Karlos RdgzNo ratings yet
- AgeLOC Power Point by Dr. Joe Chang (Aug 15, 2009)Document33 pagesAgeLOC Power Point by Dr. Joe Chang (Aug 15, 2009)trasuccess100% (1)
- Fixing Gene Expression Activity Student HandoutDocument4 pagesFixing Gene Expression Activity Student HandoutSeyeong (Daniel) NamkoongNo ratings yet
- Darren Snellgrove - Pharmaceutical MergersDocument30 pagesDarren Snellgrove - Pharmaceutical MergersSohaib TNo ratings yet
- Chromatin Accessibility Dynamics During Chemical Induction of PL 20181Document20 pagesChromatin Accessibility Dynamics During Chemical Induction of PL 20181Benn BasilNo ratings yet
- Biochemistry PowerpointDocument14 pagesBiochemistry PowerpointLelaNo ratings yet
- 2020 03 31 016972v2 FullDocument123 pages2020 03 31 016972v2 FullEmma LouthNo ratings yet
- Vaccination CertificateDocument1 pageVaccination CertificateAjay MahtoNo ratings yet
- SikimatDocument9 pagesSikimatsilviaNo ratings yet
- The EMBL Nucleotide Sequence DatabaseDocument5 pagesThe EMBL Nucleotide Sequence DatabaseJorge Luis ParraNo ratings yet
- List of Biotech Companies in Mumbai PDFDocument17 pagesList of Biotech Companies in Mumbai PDFaniket100% (1)
- Module 6 SAS Original - Docx 1Document12 pagesModule 6 SAS Original - Docx 1jenet soleilNo ratings yet
- W2-10 Overview of Amino Acid Metabolism and Protein Degradation LectureDocument31 pagesW2-10 Overview of Amino Acid Metabolism and Protein Degradation LectureSAHIL AGARWALNo ratings yet
- Bio SafetyDocument2 pagesBio Safetymalenya1100% (1)
- Chapter 3 Use of Bioinformatics in Planning A Protein 2009 Methods in EnzymDocument8 pagesChapter 3 Use of Bioinformatics in Planning A Protein 2009 Methods in Enzym王少康No ratings yet
- ParticipantCaseWorksheets 072018Document11 pagesParticipantCaseWorksheets 072018Saul0% (7)
- Topic 4 Genetic Control My NotesDocument51 pagesTopic 4 Genetic Control My NotesrukashablessingNo ratings yet