You might also like
- MCAT Content MasterlistDocument24 pagesMCAT Content MasterlistTrang LamNo ratings yet
- 21 Selection and Speciation-SDocument9 pages21 Selection and Speciation-SpyamupomNo ratings yet
- HomolgyDocument21 pagesHomolgySatish SahuNo ratings yet
- Homology ModellingDocument21 pagesHomology ModellingJerome FrancisNo ratings yet
- Pre-Assessment QuestionsDocument18 pagesPre-Assessment Questionsk.sachinNo ratings yet
- Homology ModelingDocument22 pagesHomology ModelingBasab GhoshNo ratings yet
- Homologymodelingmodeller 181002182751Document19 pagesHomologymodelingmodeller 181002182751Jhanvi SNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
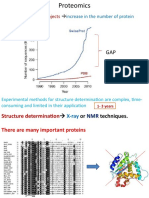
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Protein Structure Prediction TechniquesDocument30 pagesProtein Structure Prediction TechniquesJhanvi SNo ratings yet
- 3D Structure of Proteins Prediction MethodsDocument9 pages3D Structure of Proteins Prediction MethodsladykikyoyunaNo ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- Homology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersDocument16 pagesHomology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersPallavi VermaNo ratings yet
- Homology Modeling Technique for Protein Structure PredictionDocument19 pagesHomology Modeling Technique for Protein Structure PredictionJainendra JainNo ratings yet
- Homologymodeling 150123025144 Conversion Gate01Document27 pagesHomologymodeling 150123025144 Conversion Gate01Jhanvi SNo ratings yet
- Protein Tertiaty Structure PredictionDocument12 pagesProtein Tertiaty Structure PredictionRoshan Mammen ChakkungalNo ratings yet
- Homology Modelling: A 5-Step ProcessDocument30 pagesHomology Modelling: A 5-Step ProcesskamleshNo ratings yet
- Homology Modelling: Structure Prediction Using TemplatesDocument19 pagesHomology Modelling: Structure Prediction Using TemplatesManoharNo ratings yet
- 3D Structure PredictionDocument33 pages3D Structure PredictionAyesha KhanNo ratings yet
- Re Comb 2003Document16 pagesRe Comb 2003mustafaNo ratings yet
- Homology ModellingDocument29 pagesHomology ModellingRISHITA CHAUHANNo ratings yet
- 791 Ak Lecture6Document50 pages791 Ak Lecture6srikalyani2k9No ratings yet
- Protein Modelling MethodsDocument19 pagesProtein Modelling MethodsrednriNo ratings yet
- Protein ModelingDocument17 pagesProtein ModelingManeeshAgrawalNo ratings yet
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- Structural BioinfoDocument76 pagesStructural BioinfoMudit MisraNo ratings yet
- 10 21105 Joss 05057Document5 pages10 21105 Joss 05057Arpanet ArpanetNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- I-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDocument14 pagesI-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDavidNo ratings yet
- 2010 Proteins 78 118-134Document17 pages2010 Proteins 78 118-134mbrylinskiNo ratings yet
- SHARPEN Software for Protein Structure Prediction and DesignDocument10 pagesSHARPEN Software for Protein Structure Prediction and DesignSuchismita PatraNo ratings yet
- Generative Modeling for Protein StructuresDocument12 pagesGenerative Modeling for Protein Structurescarlos ArozamenaNo ratings yet
- CSC8312 Revision Lecture Exam Format and TipsDocument30 pagesCSC8312 Revision Lecture Exam Format and TipsUday KiranNo ratings yet
- Cavidad PrediccionDocument9 pagesCavidad PrediccionLuis MartinezNo ratings yet
- Protein Structure: Predictive Methods and Experimental MethodologiesDocument33 pagesProtein Structure: Predictive Methods and Experimental MethodologiesmarylaranjoNo ratings yet
- Bioinfo - S1 2021 - L9 - Protein Structure - 1 SlideDocument87 pagesBioinfo - S1 2021 - L9 - Protein Structure - 1 SlideHuynh Ngoc Da ThaoNo ratings yet
- Identification of Modelling TemplatesDocument7 pagesIdentification of Modelling TemplatesLokesh AdhikariNo ratings yet
- Automated Design of The Surface Positions of Protein HelicesDocument9 pagesAutomated Design of The Surface Positions of Protein Helicesaxva1663No ratings yet
- Structural Biology: What Does 3D Tell Us?Document20 pagesStructural Biology: What Does 3D Tell Us?Prakash KumarNo ratings yet
- Methods For Accurate Homology Modeling by Global OptimizationDocument17 pagesMethods For Accurate Homology Modeling by Global OptimizationUmmuNo ratings yet
- Proteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingDocument10 pagesProteins: Benchmarking Template Selection and Model Quality Assessment For High-Resolution Comparative ModelingLavanya Priya SathyanNo ratings yet
- Proteins Bioinfo LatestDocument45 pagesProteins Bioinfo Latestpeace loverNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Protein Structure Prediction ThesisDocument8 pagesProtein Structure Prediction ThesisWriteMyThesisPaperManchester100% (2)
- Protein Structure Prediction ToolsDocument63 pagesProtein Structure Prediction ToolsAnjana's WorldNo ratings yet
- Fold LibDocument24 pagesFold LibArnish Chakraborty100% (1)
- Cyclic peptide structure prediction and design using AlphaFoldDocument25 pagesCyclic peptide structure prediction and design using AlphaFoldxinyuwu0528No ratings yet
- Bif501 AssDocument3 pagesBif501 Asssana 568aNo ratings yet
- Sanchez CurrOpinStructBiol 1997Document9 pagesSanchez CurrOpinStructBiol 1997Phoenix RisingNo ratings yet
- Biopolymer: Structural Biology: Macromolecular ModelingDocument4 pagesBiopolymer: Structural Biology: Macromolecular Modelingalusia24No ratings yet
- Simulation Based Drug Discovery TechniquesDocument31 pagesSimulation Based Drug Discovery TechniquesBagya VetriNo ratings yet
- Struct GenomDocument7 pagesStruct GenomNaidelyn HernandezNo ratings yet
- Protein Structural Motifs: Doug Brutlag Professor Emeritus Biochemistry & Medicine (By Courtesy)Document100 pagesProtein Structural Motifs: Doug Brutlag Professor Emeritus Biochemistry & Medicine (By Courtesy)Joan Manuel Tovar CasallasNo ratings yet
- 2011 J Struct Biol 173 558-569Document12 pages2011 J Struct Biol 173 558-569mbrylinskiNo ratings yet
- 7 HomologyModelling 12oct2020Document8 pages7 HomologyModelling 12oct2020things strangerNo ratings yet
- Protein ModellingDocument15 pagesProtein ModellingNandini KotharkarNo ratings yet
- MP-T Improving Membrane Protein Alignment For StructureDocument8 pagesMP-T Improving Membrane Protein Alignment For StructureCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Improved Prediction of Protein Side-Chain Conformations With SCWRL4Document18 pagesImproved Prediction of Protein Side-Chain Conformations With SCWRL4dunbrackNo ratings yet
- Prediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsDocument11 pagesPrediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsLata DeshmukhNo ratings yet
- Protein Modeling: Protein Structure Prediction Other TopicsDocument76 pagesProtein Modeling: Protein Structure Prediction Other Topicsuma-chenNo ratings yet
- Insilico DockingDocument6 pagesInsilico DockingRajesh GuruNo ratings yet
- Biomolecular Networks: Methods and Applications in Systems BiologyFrom EverandBiomolecular Networks: Methods and Applications in Systems BiologyNo ratings yet
- OISB PDF Silver StainingDocument2 pagesOISB PDF Silver Stainingsurrender003No ratings yet
- Complete General Science For NSEJS NTSE KVPY IAS Physics Chemistry Biology Sifox Publication Praveen Kumar PDFDocument550 pagesComplete General Science For NSEJS NTSE KVPY IAS Physics Chemistry Biology Sifox Publication Praveen Kumar PDFtanisha pianoNo ratings yet
- Somogyi NelsonDocument6 pagesSomogyi NelsonPhichamon NoisuwanNo ratings yet
- Burden of Skin Diseases 2004Document110 pagesBurden of Skin Diseases 2004surrender003No ratings yet
- Nelson and SomogyiDocument3 pagesNelson and Somogyisurrender003No ratings yet
- Nelson and SomogyiDocument3 pagesNelson and Somogyisurrender003No ratings yet
- Nelson and SomogyiDocument3 pagesNelson and Somogyisurrender003No ratings yet
- Cancer Cell Development - Lecture Day 5Document17 pagesCancer Cell Development - Lecture Day 5blakeNo ratings yet
- 5 Factors Affecting Morphogenesis in VitroDocument20 pages5 Factors Affecting Morphogenesis in VitroAlec Liu0% (3)
- Branches of BiologyDocument2 pagesBranches of BiologyMia Onglao100% (5)
- Biological Science - Course OutlineDocument3 pagesBiological Science - Course Outlinetorjak shinNo ratings yet
- ExiPrep™16 BrochureDocument6 pagesExiPrep™16 BrochureNaruepon HongmanopNo ratings yet
- PCR - Real-Time QPCR - in Situ Hybridisation: Biomedical Research Techniques (MEDSCI 720)Document74 pagesPCR - Real-Time QPCR - in Situ Hybridisation: Biomedical Research Techniques (MEDSCI 720)Dr-Dalya ShakirNo ratings yet
- The Past, Present and Future of Biotechnology in MexicoDocument2 pagesThe Past, Present and Future of Biotechnology in MexicoQkriiz Arriola100% (1)
- Genetics NotesDocument6 pagesGenetics NotesShane Tang100% (2)
- Patient Information Sample Information: Test Requisition FormDocument1 pagePatient Information Sample Information: Test Requisition FormKalid Maher Al RaeeNo ratings yet
- New Insights Into The Structure and Function of Phyllosphere Microbiota Through High-Throughput Molecular ApproachesDocument10 pagesNew Insights Into The Structure and Function of Phyllosphere Microbiota Through High-Throughput Molecular ApproachesindahdwirahNo ratings yet
- European Journal of Pharmaceutical Sciences: SciencedirectDocument8 pagesEuropean Journal of Pharmaceutical Sciences: SciencedirectVania León RNo ratings yet
- DNA and RNA Structure and FunctionDocument3 pagesDNA and RNA Structure and FunctionAraquel Nunez100% (1)
- MLC Reaction Detection of Alloreactive T CellsDocument2 pagesMLC Reaction Detection of Alloreactive T CellsMuthi KhairunnisaNo ratings yet
- 2022 Book TheEBMTEHACAR-TCellHandbookDocument221 pages2022 Book TheEBMTEHACAR-TCellHandbookInchirah Narimane MgdNo ratings yet
- Impact of antimicrobial stewardship strategies on resistance and useDocument11 pagesImpact of antimicrobial stewardship strategies on resistance and useAyush KumarNo ratings yet
- General Structure and Classification of Viruses 4Document50 pagesGeneral Structure and Classification of Viruses 4OROKE JOHN EJE100% (1)
- IMNCIDocument13 pagesIMNCIJayalakshmiullasNo ratings yet
- Protein identification by peptide mass fingerprintingDocument60 pagesProtein identification by peptide mass fingerprintingNasrullahNo ratings yet
- BT102 - Microbiology (Solved Questions FINAL TERM (PAST PAPERS)Document33 pagesBT102 - Microbiology (Solved Questions FINAL TERM (PAST PAPERS)Awais BhuttaNo ratings yet
- Lecture 1 Introduction To Physiological Principles 080317Document42 pagesLecture 1 Introduction To Physiological Principles 080317api-386303659No ratings yet
- Pharmexil - All MixedDocument14 pagesPharmexil - All MixedLisa RayNo ratings yet
- Molecular Genetics RecombinationDocument55 pagesMolecular Genetics RecombinationRiyaniMartyasariNo ratings yet
- FISH protocol for formalin-fixed tissue sectionsDocument15 pagesFISH protocol for formalin-fixed tissue sectionsFlorsie MirandaNo ratings yet
- PADER TrainingDocument20 pagesPADER Trainingisha jain100% (1)
- Big Six of Developmental BiologyDocument6 pagesBig Six of Developmental BiologyLove Jade TupazNo ratings yet
- Profound Tissue Specifity in Proliferation Control Underlies Cancer Drivers and Aneuploidy PatternsDocument40 pagesProfound Tissue Specifity in Proliferation Control Underlies Cancer Drivers and Aneuploidy PatternsDAYANA ERAZONo ratings yet
- Biology: Study Package Zenith / Octagon Class XDocument19 pagesBiology: Study Package Zenith / Octagon Class Xsajal aggarwalNo ratings yet
- Allelic diversity for salt stress genes in Indian riceDocument9 pagesAllelic diversity for salt stress genes in Indian ricesalma sabilaNo ratings yet