You might also like
- Dysphagia, A Simple Guide To The Condition, Treatment And Related ConditionsFrom EverandDysphagia, A Simple Guide To The Condition, Treatment And Related ConditionsRating: 5 out of 5 stars5/5 (1)
- Cleft Lip and Palate CareDocument11 pagesCleft Lip and Palate CareEvangeline Anne MacanasNo ratings yet
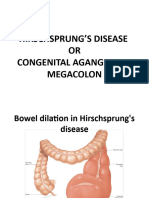
- Hirschsprung'S Disease OR Congenital Aganglionic MegacolonDocument59 pagesHirschsprung'S Disease OR Congenital Aganglionic MegacolonHarshika KDGNo ratings yet
- Gastroenteritis and Diarrhea TreatmentDocument110 pagesGastroenteritis and Diarrhea TreatmentJaja ManezNo ratings yet
- Congenital Anomalies of GiDocument94 pagesCongenital Anomalies of GiPadmaNo ratings yet
- Hirchsprung Disease: A.K.A. Congenital AganglionicDocument29 pagesHirchsprung Disease: A.K.A. Congenital Aganglionicdesh_dacanayNo ratings yet
- Paediatric Surgery 2Document38 pagesPaediatric Surgery 2عمار عارفNo ratings yet
- University of Mosul / College of Nursing Child and Adolescent Health NursingDocument11 pagesUniversity of Mosul / College of Nursing Child and Adolescent Health NursingRayan AhmedNo ratings yet
- Pedia GiDocument10 pagesPedia GiCam IbardolasaNo ratings yet
- Abdominal Wall Defects: Omphalocele and Gastroschisis: DR - Enono Yhoshu Department of Pediatric SurgeryDocument42 pagesAbdominal Wall Defects: Omphalocele and Gastroschisis: DR - Enono Yhoshu Department of Pediatric SurgeryYogi drNo ratings yet
- Hirsch SprungDocument3 pagesHirsch SprungBheru LalNo ratings yet
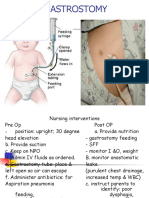
- GASTROSTOMY FEEDING CAREDocument26 pagesGASTROSTOMY FEEDING CAREAileen MonaresNo ratings yet
- Intussuseption and Hirschprung's DiseaseDocument5 pagesIntussuseption and Hirschprung's DiseaseAris Magallanes100% (2)
- Intussusception: Prepared By: Aisha H. AlaghaDocument20 pagesIntussusception: Prepared By: Aisha H. AlaghaaiooshaNo ratings yet
- Cleft Lip and Palate Guide: Causes, Treatment and MoreDocument116 pagesCleft Lip and Palate Guide: Causes, Treatment and MoremeleseNo ratings yet
- Askep HisprungDocument25 pagesAskep HisprungRika AmaliyaNo ratings yet
- Digestive DiseasesDocument10 pagesDigestive Diseaseskate_5182178No ratings yet
- Cleft Lip / Cleft Palate: Predisposing FactorsDocument15 pagesCleft Lip / Cleft Palate: Predisposing FactorsMabesNo ratings yet
- Pyloric Stenosis WDocument11 pagesPyloric Stenosis WKlaue Neiv CallaNo ratings yet
- No Bowel Output in NeonatesDocument24 pagesNo Bowel Output in NeonatesOTOH RAYA OMARNo ratings yet
- 8. Care of Child With GI Dysfunction (1) ءءءءDocument44 pages8. Care of Child With GI Dysfunction (1) ءءءءNuhaNo ratings yet
- Defecte Abdominale NNDocument56 pagesDefecte Abdominale NNDiana CostanNo ratings yet
- 2 Common Congenital Anomalies of The GI TractDocument27 pages2 Common Congenital Anomalies of The GI TractZyke NovenoNo ratings yet
- Imperforate AnusDocument17 pagesImperforate AnusNalzaro Emyril100% (1)
- Hirschsprung Disease: Causes, Symptoms and Treatment/TITLEDocument61 pagesHirschsprung Disease: Causes, Symptoms and Treatment/TITLEAdditi Satyal100% (1)
- Hernia: Under Supervision DR Arzak SaberDocument32 pagesHernia: Under Supervision DR Arzak Sabermathio medhatNo ratings yet
- GI DisordersDocument42 pagesGI Disordersvarshasharma05No ratings yet
- Hirschsprung Disease: Causes, Symptoms, Diagnosis and TreatmentDocument10 pagesHirschsprung Disease: Causes, Symptoms, Diagnosis and TreatmentUday KumarNo ratings yet
- Lecture 1 HirschprungDocument18 pagesLecture 1 HirschprungsharmeenNo ratings yet
- Patient Scenario, Chapter 45, Nursing Care of A Family When A Child Has A Gastrointestinal DisorderDocument93 pagesPatient Scenario, Chapter 45, Nursing Care of A Family When A Child Has A Gastrointestinal DisorderDay MedsNo ratings yet
- Pyloric Stenosis: Supervisor: DR NTAGANDA Edmond, Consultant Ped SurgDocument21 pagesPyloric Stenosis: Supervisor: DR NTAGANDA Edmond, Consultant Ped SurgJames NTEGEREJIMANANo ratings yet
- Congenital Anomalies: Prof Mrs. M. Radhika HOD, Dept of Nursing Research Narayana College of Nursing NelloreDocument73 pagesCongenital Anomalies: Prof Mrs. M. Radhika HOD, Dept of Nursing Research Narayana College of Nursing NelloreSAMHITHA JNo ratings yet
- Neonatal Anaesthesia 2 Anaesthesia For Neonates With Abdominal Wall DefectsDocument10 pagesNeonatal Anaesthesia 2 Anaesthesia For Neonates With Abdominal Wall DefectsDr. Anish GolchhaNo ratings yet
- Emergency management of constipation in childrenDocument10 pagesEmergency management of constipation in childrenAlma Rebeca Mota NovaNo ratings yet
- In Partial Fulfillment For The Requirements in R.L.E: Submitted By: Elaine Blanche R. GonzalezDocument6 pagesIn Partial Fulfillment For The Requirements in R.L.E: Submitted By: Elaine Blanche R. GonzalezBlanche GonzalezNo ratings yet
- Gastro NephroDocument93 pagesGastro Nephrohasanatiya41No ratings yet
- Competency Appraisal 1 Reviewer-1Document53 pagesCompetency Appraisal 1 Reviewer-1Paolo Carubio100% (1)
- Foreign Body Ingestion: Leano, Fides Beatrice D.LDocument29 pagesForeign Body Ingestion: Leano, Fides Beatrice D.L3sshhhNo ratings yet
- Pedia PT3Document8 pagesPedia PT3sonava vichNo ratings yet
- HIRSCHSPRUNG - Surgical Presentation HSCDocument34 pagesHIRSCHSPRUNG - Surgical Presentation HSCKimkimkim Eligio100% (1)
- SG3 Paediatric Surgical EmergenciesDocument69 pagesSG3 Paediatric Surgical EmergenciesDiyana ZatyNo ratings yet
- How to Do Hospital Audit Beginners GuideDocument46 pagesHow to Do Hospital Audit Beginners GuideKASATSANo ratings yet
- Pyloric Stenosis Case StudyDocument37 pagesPyloric Stenosis Case StudyFaith Torralba100% (3)
- Hisprung DiseaseDocument12 pagesHisprung DiseaseEky Madyaning NastitiNo ratings yet
- Pyloric Stenosis: CLASS:-B.Sc. Nursing 3 YearDocument38 pagesPyloric Stenosis: CLASS:-B.Sc. Nursing 3 YearshikhaNo ratings yet
- Pyloric Stenosis: CLASS:-B.Sc. Nursing 3 YearDocument38 pagesPyloric Stenosis: CLASS:-B.Sc. Nursing 3 Yearshikha100% (1)
- Esophageal AtresiaDocument18 pagesEsophageal AtresiaNeha RathoreNo ratings yet
- Abdominal Wall Defects PresentationDocument45 pagesAbdominal Wall Defects PresentationWasswaNo ratings yet
- Infant GI Disorders GuideDocument80 pagesInfant GI Disorders GuidejoycechicagoNo ratings yet
- MiniOSCE Surgery 1Document329 pagesMiniOSCE Surgery 1Mohammad BanisalmanNo ratings yet
- Gastrointestinal Issues in ChildrenDocument10 pagesGastrointestinal Issues in ChildrenANNENo ratings yet
- IntussusceptionDocument10 pagesIntussusceptionshenlie100% (1)
- Gastrointestinal Fistula GuideDocument60 pagesGastrointestinal Fistula GuideSangija kamataNo ratings yet
- Chapter 28 Child With A Gastrointestinal ConditionDocument85 pagesChapter 28 Child With A Gastrointestinal Conditionsey_scottNo ratings yet
- Pediatric G.I Disorders FinalDocument53 pagesPediatric G.I Disorders FinalRashid Hussain0% (1)
- Pediatric Care Online AAPDocument18 pagesPediatric Care Online AAPNikola StojsicNo ratings yet
- Genital ProlapseDocument20 pagesGenital ProlapseMehreen MaqboolNo ratings yet
- Infantile Hypertrophic Pyloric Stenosis (IHPS) : IncidenceDocument9 pagesInfantile Hypertrophic Pyloric Stenosis (IHPS) : IncidenceRaed AlhnaityNo ratings yet
- 3 Common Pediatric Surgery ContinuedDocument5 pages3 Common Pediatric Surgery ContinuedMohamed Al-zichrawyNo ratings yet
- Pyloric stenosis guide: causes, symptoms, diagnosis and treatmentDocument5 pagesPyloric stenosis guide: causes, symptoms, diagnosis and treatmentfdgrgNo ratings yet
- Taibah University Nursing Case Discussion EvaluationDocument1 pageTaibah University Nursing Case Discussion Evaluationمهند الرحيليNo ratings yet
- 6 Bedside & NCP Discussion Evaluation RubricDocument2 pages6 Bedside & NCP Discussion Evaluation Rubricمهند الرحيليNo ratings yet
- Geriatric Assessment SheetDocument14 pagesGeriatric Assessment Sheetمهند الرحيليNo ratings yet
- Taibah University College of Nursing drug study rubricDocument2 pagesTaibah University College of Nursing drug study rubricزياد- 90No ratings yet
- Nursing Care Plan (NCP) : Patient-Centered (In Priority Order)Document1 pageNursing Care Plan (NCP) : Patient-Centered (In Priority Order)مهند الرحيليNo ratings yet
- 4 Drug Study FormDocument1 page4 Drug Study Formمهند الرحيليNo ratings yet
- Lecture 7 Cardiovascular Dysfunction, Altered Fluid Balance and Mal Nutrition 2021 2Document57 pagesLecture 7 Cardiovascular Dysfunction, Altered Fluid Balance and Mal Nutrition 2021 2مهند الرحيليNo ratings yet
- Lecture 8, Nursing Care of The Child With Respiratory Dysfunction and Renal Disorders 2Document48 pagesLecture 8, Nursing Care of The Child With Respiratory Dysfunction and Renal Disorders 2مهند الرحيليNo ratings yet
- 9 - Antiprotozoal DrugsDocument29 pages9 - Antiprotozoal Drugsمهند الرحيليNo ratings yet
- 8 - Antiviral DrugsDocument29 pages8 - Antiviral Drugsمهند الرحيليNo ratings yet
- Week 2 Pedia Res, FeedDocument58 pagesWeek 2 Pedia Res, Feedمهند الرحيليNo ratings yet
- Perspectives of Paediatric NursingDocument35 pagesPerspectives of Paediatric Nursingمهند الرحيليNo ratings yet
- Lecture 8, Nursing Care of The Child With Respiratory Dysfunction and Renal Disorders 2Document48 pagesLecture 8, Nursing Care of The Child With Respiratory Dysfunction and Renal Disorders 2مهند الرحيليNo ratings yet
- Growth & Development and Developmental TheoryDocument46 pagesGrowth & Development and Developmental Theoryمهند الرحيليNo ratings yet
- Lecture 7 Cardiovascular Dysfunction, Altered Fluid Balance and Mal Nutrition 2021 2Document57 pagesLecture 7 Cardiovascular Dysfunction, Altered Fluid Balance and Mal Nutrition 2021 2مهند الرحيليNo ratings yet
- Week 2 Pedia Res, FeedDocument58 pagesWeek 2 Pedia Res, Feedمهند الرحيليNo ratings yet
- Perspectives of Paediatric NursingDocument35 pagesPerspectives of Paediatric Nursingمهند الرحيليNo ratings yet
- Growth and Development of School ChildrenDocument44 pagesGrowth and Development of School Childrenمهند الرحيليNo ratings yet
- New Born, Infant and ToddlerDocument61 pagesNew Born, Infant and Toddlerمهند الرحيليNo ratings yet
- Growth & Development and Developmental TheoryDocument46 pagesGrowth & Development and Developmental Theoryمهند الرحيليNo ratings yet
- Pediatric Nursing - Newborn Assessment & Growth DevelopmentDocument5 pagesPediatric Nursing - Newborn Assessment & Growth DevelopmentRegieLimos100% (2)
- Cleft Lip and PalateDocument76 pagesCleft Lip and PalateMira Hazwani100% (1)
- The Effects of Lactation Education and A Prosthetic Obturator Appliance On Feeding Efficiency in Infants With Cleft Lip and PalateDocument6 pagesThe Effects of Lactation Education and A Prosthetic Obturator Appliance On Feeding Efficiency in Infants With Cleft Lip and PalateingridspulerNo ratings yet
- Lesson Plan On KatoriDocument11 pagesLesson Plan On KatoriNimi SusanthomasNo ratings yet
- Posters FinalDocument70 pagesPosters FinalShkelzen KomoniNo ratings yet
- Velopharyngeal InsufficiencyDocument9 pagesVelopharyngeal InsufficiencyJASPREETKAUR0410No ratings yet
- Week 4 MCQDocument30 pagesWeek 4 MCQZainab_Wajih_2544100% (1)
- Dental LabDocument109 pagesDental Labvinay kumarNo ratings yet
- CLCPDocument43 pagesCLCPeviltohuntNo ratings yet
- Trainer Manual For Nusring Care Saves LivesDocument15 pagesTrainer Manual For Nusring Care Saves LivesEko AwaluddinNo ratings yet
- Developmental Disturbances of Oral and Paraoral StructuresDocument14 pagesDevelopmental Disturbances of Oral and Paraoral StructuresYashmeen XNo ratings yet
- Cleft Lip and Palate Treatment HistoryDocument6 pagesCleft Lip and Palate Treatment HistoryArchanaShenoyNo ratings yet
- Written Assignment 1Document5 pagesWritten Assignment 1api-401934380No ratings yet
- Preventative Partner Safety AuditDocument23 pagesPreventative Partner Safety AuditINSTALASI BEDAH SENTRALNo ratings yet
- Oral and Maxillofacial Pathology (Nuevo)Document986 pagesOral and Maxillofacial Pathology (Nuevo)Leonel Larregui83% (23)
- Cone Beam Tomography in Orthodontics PDFDocument8 pagesCone Beam Tomography in Orthodontics PDFdruzair007No ratings yet
- Respiratory AssessmentDocument25 pagesRespiratory AssessmentchristyNo ratings yet
- Cleft Lip & Palate Embryology, Causes, ClassificationDocument46 pagesCleft Lip & Palate Embryology, Causes, ClassificationDr.Neethu Salam100% (3)
- LABIOPALATOSCHIZISDocument25 pagesLABIOPALATOSCHIZISEya Prepti SerraNo ratings yet
- Earproblemsinchildren WithcleftpalatesDocument3 pagesEarproblemsinchildren Withcleftpalatesane_dokter1905No ratings yet
- Cleft Lip and PalateDocument38 pagesCleft Lip and PalateHarry Sudarma AmersonNo ratings yet
- Hyperthermia and Risk For AspirationDocument3 pagesHyperthermia and Risk For AspirationAlmyr RimandoNo ratings yet
- Neville's Atlas of Oral Pathology - TEXTODocument488 pagesNeville's Atlas of Oral Pathology - TEXTOgagandeep singh100% (1)
- Cleft Lip and Palate Surgery: The Day of SurgeryDocument4 pagesCleft Lip and Palate Surgery: The Day of SurgeryBean GemboelzNo ratings yet
- ENT 1.3 Lips and Oral CavityDocument15 pagesENT 1.3 Lips and Oral CavityZazaNo ratings yet
- Nasal EmbryoDocument6 pagesNasal EmbryoLevani KartvelishviliNo ratings yet
- New Born Assess PPT FrankDocument118 pagesNew Born Assess PPT FrankPooja HardiyaNo ratings yet
- Cleft Lip and PalateDocument34 pagesCleft Lip and PalateatikaNo ratings yet
- 3 (OB Cases)Document267 pages3 (OB Cases)Sharyl Plan SarominesNo ratings yet
- Zofran 1Document10 pagesZofran 1api-301957730No ratings yet