You might also like
- Medicine in Brief: Name the Disease in Haiku, Tanka and ArtFrom EverandMedicine in Brief: Name the Disease in Haiku, Tanka and ArtRating: 5 out of 5 stars5/5 (1)
- Tetralogy of FallotDocument28 pagesTetralogy of FallotArdyansyah Nasution, M.DNo ratings yet
- Tetralogy of Fallot Lesson PlanDocument25 pagesTetralogy of Fallot Lesson PlanUday Kumar100% (1)
- Guide to Basic and Extended FATE ViewsDocument4 pagesGuide to Basic and Extended FATE ViewsMateen Shukri100% (2)
- Hasil Tesis AdelaideDocument93 pagesHasil Tesis AdelaideAndi Alfian Zainuddin100% (1)
- Presentation ON Tetrallogy of FallotDocument33 pagesPresentation ON Tetrallogy of Fallotvani reddyNo ratings yet
- Odia Tetralogy of FallotDocument25 pagesOdia Tetralogy of Fallotvictorjonathan567No ratings yet
- Tetralogy of Fallot: History & ExamDocument35 pagesTetralogy of Fallot: History & ExamMicija CucuNo ratings yet
- 1. Tetralogy of Fallot - StatPearls - NCBI BookshelfDocument9 pages1. Tetralogy of Fallot - StatPearls - NCBI BookshelfElfrida AuliaNo ratings yet
- Tetralogy of FallotDocument65 pagesTetralogy of FallotEndrati WidiastutiNo ratings yet
- Tetralogy of FallotDocument7 pagesTetralogy of FallotAshok Kumar JangirNo ratings yet
- Tetralogy of Fallot ExplainedDocument31 pagesTetralogy of Fallot ExplainedDevipriya MajumderNo ratings yet
- Tetralogy of FallotDocument28 pagesTetralogy of FallotconcozNo ratings yet
- Tetralogy of FallotDocument7 pagesTetralogy of FallotWidelmark FarrelNo ratings yet
- LESSON 3. Congenital Heart DefectsDocument9 pagesLESSON 3. Congenital Heart DefectsEdmarie P. TabacoldeNo ratings yet
- Tetralogy of FallotDocument31 pagesTetralogy of FallotAnditha Namira RS100% (1)
- Tetralogy of Fallot ExplainedDocument9 pagesTetralogy of Fallot ExplainedLaurice BarronNo ratings yet
- Case Report: Department of Cardiology, IRCCS San Raffaele Hospital, Milan, ItalyDocument4 pagesCase Report: Department of Cardiology, IRCCS San Raffaele Hospital, Milan, ItalyAndreas Erick HaurissaNo ratings yet
- Echocardiographic Evaluation of Tetralogy of Fallot : Echo in Adult Congenital Heart DiseaseDocument9 pagesEchocardiographic Evaluation of Tetralogy of Fallot : Echo in Adult Congenital Heart DiseaseResiden KardiologiNo ratings yet
- TOF Heart Defect Guide: Symptoms, Causes, Diagnosis & TreatmentDocument8 pagesTOF Heart Defect Guide: Symptoms, Causes, Diagnosis & Treatmentdecembergirl92No ratings yet
- Cardiology Best RDocument11 pagesCardiology Best RislamawniNo ratings yet
- Understanding Tetralogy of Fallot: Causes, Symptoms and TreatmentDocument7 pagesUnderstanding Tetralogy of Fallot: Causes, Symptoms and TreatmentJoanna Bee Rose MagyawiNo ratings yet
- TOF Pathophysiology ExplainedDocument30 pagesTOF Pathophysiology ExplainedArianna Jasmine MabungaNo ratings yet
- Pedia Ward Case ConDocument13 pagesPedia Ward Case ConAnthony Arcenal TarnateNo ratings yet
- Cardiac Effusions: Causes, Symptoms and DiagnosisDocument26 pagesCardiac Effusions: Causes, Symptoms and DiagnosisjsenocNo ratings yet
- Tetralogy of Fallot SP B.inggrisDocument19 pagesTetralogy of Fallot SP B.inggrisSoraya VerinaNo ratings yet
- Tetralogy of FallotDocument24 pagesTetralogy of FallotjustinahorroNo ratings yet
- Diagnosis and Management Issues in Thoracic Aortic AneurysmDocument10 pagesDiagnosis and Management Issues in Thoracic Aortic AneurysmLisa TuneNo ratings yet
- Tetralogy of Fallot, Agarwala 2017Document5 pagesTetralogy of Fallot, Agarwala 2017rinayondaNo ratings yet
- Fallot TetralogyDocument20 pagesFallot Tetralogykgt88No ratings yet
- Mac Donald 2011Document17 pagesMac Donald 2011Paola ChanNo ratings yet
- Approach To A Child With Congenital Heart DiseaseDocument31 pagesApproach To A Child With Congenital Heart DiseaseMUHAMMAD DANIAL BIN HASAN FPSKNo ratings yet
- Tetralogy of FallotDocument9 pagesTetralogy of FallotPaul MagbooNo ratings yet
- Tetralogy of Fallot Report 1Document9 pagesTetralogy of Fallot Report 1Maricar EustaquioNo ratings yet
- Congenital Heart Defec1Document3 pagesCongenital Heart Defec1Matt BaterzalNo ratings yet
- Tetralogy of Fallot Everything You Wanted To Know But Were Afraid To AskDocument8 pagesTetralogy of Fallot Everything You Wanted To Know But Were Afraid To AsksofiaNo ratings yet
- Tetralogy of Fallot: Everything You Wanted To Know But Were Afraid To AskDocument8 pagesTetralogy of Fallot: Everything You Wanted To Know But Were Afraid To AskTuong NguyenNo ratings yet
- Congenital Heart Disease (2011)Document17 pagesCongenital Heart Disease (2011)drheay100% (1)
- Ciano Tico IDocument14 pagesCiano Tico ILilik FitrianaNo ratings yet
- Pulmonary hypertension causes and symptomsDocument10 pagesPulmonary hypertension causes and symptomscharmieangelNo ratings yet
- Tetralogy of Fallot Cyanotic Lesion GuideDocument12 pagesTetralogy of Fallot Cyanotic Lesion Guideputri nadNo ratings yet
- Barlows Syndrome PPT AbhishekDocument10 pagesBarlows Syndrome PPT AbhishekAbhishek SagarNo ratings yet
- Congenital heart defect guideDocument30 pagesCongenital heart defect guideIshaan KumarNo ratings yet
- Case Study Worksheet EndocarditisDocument6 pagesCase Study Worksheet EndocarditisSharlee StoneNo ratings yet
- Filamin A Mutations: Jesse Hansen-BartelDocument8 pagesFilamin A Mutations: Jesse Hansen-BartelJesse Helmut Hansen-BartelNo ratings yet
- Primary Pulmonary Hypertension PathophysiologyDocument14 pagesPrimary Pulmonary Hypertension PathophysiologygibreilNo ratings yet
- Cayanotic Heart DiseaseDocument28 pagesCayanotic Heart DiseasesivanathanNo ratings yet
- Tetralogy of FallotDocument22 pagesTetralogy of FallotMawaddah NurrahmaNo ratings yet
- Abstract TOFDocument1 pageAbstract TOFUlfia SafitriNo ratings yet
- Management of Heart Failure: DR Ambakederemo TE Consultant Physician/cardiologist NduthDocument71 pagesManagement of Heart Failure: DR Ambakederemo TE Consultant Physician/cardiologist NduthPrincewill SeiyefaNo ratings yet
- Tetralogy of Fallot.......Document7 pagesTetralogy of Fallot.......Sam AlipioNo ratings yet
- Venous Thromboembolic DiseaseDocument27 pagesVenous Thromboembolic DiseaseAndra BauerNo ratings yet
- Congenital Heart Diseases.Document86 pagesCongenital Heart Diseases.Salman KhanNo ratings yet
- Tetralogy of Fallot with Hypercyanotic SpellsDocument11 pagesTetralogy of Fallot with Hypercyanotic SpellsPatrick DycocoNo ratings yet
- 1.3 Heart FailureDocument30 pages1.3 Heart Failuresn_b5No ratings yet
- Tetralogy of FallotDocument3 pagesTetralogy of FallotMiLody NajeNo ratings yet
- Pda TofDocument56 pagesPda TofPritam PanigrahiNo ratings yet
- Tetralogy of FallotDocument5 pagesTetralogy of FallotSaloni MehtaNo ratings yet
- B19 Pleno Sken2Document15 pagesB19 Pleno Sken2Adelia YuantikaNo ratings yet
- Cardio Vascular DisordersDocument62 pagesCardio Vascular DisordersUday Kumar100% (1)
- Congenital Heart Diseases, A Simple Guide to these Medical ConditionsFrom EverandCongenital Heart Diseases, A Simple Guide to these Medical ConditionsNo ratings yet
- DiabetesDocument47 pagesDiabetesvani reddyNo ratings yet
- Cystic FibrosisDocument35 pagesCystic Fibrosisvani reddyNo ratings yet
- CardiologyDocument29 pagesCardiologyvani reddyNo ratings yet
- Inservice EducatrionDocument28 pagesInservice Educatrionvani reddyNo ratings yet
- Types of FeverDocument10 pagesTypes of Fevervani reddyNo ratings yet
- Ward TeachingDocument23 pagesWard Teachingvani reddyNo ratings yet
- Child DevelopmentDocument7 pagesChild Developmentvani reddyNo ratings yet
- Problems Faced by Maternal and Child Health Nursing UnitsDocument19 pagesProblems Faced by Maternal and Child Health Nursing Unitsvani reddyNo ratings yet
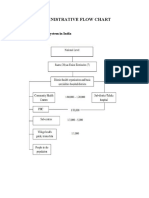
- Administrative Flow Chart: Health Care System in IndiaDocument4 pagesAdministrative Flow Chart: Health Care System in Indiavani reddyNo ratings yet
- Government College of Nursing. Somajiguda, HyderabadDocument1 pageGovernment College of Nursing. Somajiguda, Hyderabadvani reddyNo ratings yet
- The Referral System Revised: DisclaimerDocument33 pagesThe Referral System Revised: Disclaimervani reddyNo ratings yet
- Problem Solving Approach Topic WordDocument20 pagesProblem Solving Approach Topic Wordvani reddyNo ratings yet
- Management ClinicalsDocument11 pagesManagement Clinicalsvani reddyNo ratings yet
- Government College of Nursing List of Bank Details of MSC (N) 2 YEAR STUDENTS (2019-2021)Document1 pageGovernment College of Nursing List of Bank Details of MSC (N) 2 YEAR STUDENTS (2019-2021)vani reddyNo ratings yet
- Jhpiego Pinktober OutlineDocument1 pageJhpiego Pinktober Outlinevani reddyNo ratings yet
- Bone Cancer and Bone Infection Diagnosis and TreatmentDocument85 pagesBone Cancer and Bone Infection Diagnosis and Treatmentvani reddyNo ratings yet
- Overview of Cystic Fibrosis & Its ManagementDocument54 pagesOverview of Cystic Fibrosis & Its Managementvani reddyNo ratings yet
- Problem Solving Approach Topic WordDocument20 pagesProblem Solving Approach Topic Wordvani reddyNo ratings yet
- Government College of Nursing List of Bank Details of MSC (N) 2 YEAR STUDENTS (2019-2021)Document1 pageGovernment College of Nursing List of Bank Details of MSC (N) 2 YEAR STUDENTS (2019-2021)vani reddyNo ratings yet
- Jhpiego Pinktober OutlineDocument1 pageJhpiego Pinktober Outlinevani reddyNo ratings yet
- Ludwing and Voin Bertalanffys General System TheoryDocument1 pageLudwing and Voin Bertalanffys General System Theoryvani reddyNo ratings yet
- Government College of Nursing MSC (N) 1 ST YEAR (2019-2021) Research Proposal PresentationsDocument4 pagesGovernment College of Nursing MSC (N) 1 ST YEAR (2019-2021) Research Proposal Presentationsvani reddyNo ratings yet
- Presentation Title 2/11/20XXDocument18 pagesPresentation Title 2/11/20XXvani reddyNo ratings yet
- ANTIPYRETICSDocument34 pagesANTIPYRETICSvani reddyNo ratings yet
- Cystic Fibrosis: A Lethal Autosomal Recessive Disease Affecting Multiple OrgansDocument16 pagesCystic Fibrosis: A Lethal Autosomal Recessive Disease Affecting Multiple Organsvani reddyNo ratings yet
- Government College of Nursing MSC (N) 1 ST YEAR (2019-2021) Research Proposal PresentationsDocument4 pagesGovernment College of Nursing MSC (N) 1 ST YEAR (2019-2021) Research Proposal Presentationsvani reddyNo ratings yet
- Lesson Plan ON BroncitisDocument24 pagesLesson Plan ON Broncitisvani reddyNo ratings yet
- Vice President Maria Leonor Leni Gerona RobredoDocument2 pagesVice President Maria Leonor Leni Gerona RobredoMikaela Marie Dela CruzNo ratings yet
- Cor Pulmonale ReDocument35 pagesCor Pulmonale ReFabb NelsonNo ratings yet
- Heart Failure and Ejection FractionDocument1 pageHeart Failure and Ejection FractionCecil-An DalanonNo ratings yet
- CVS TimetableDocument6 pagesCVS TimetableKai BinNo ratings yet
- Cardiac Emergencies GuideDocument49 pagesCardiac Emergencies Guideraman kumari100% (3)
- Science Activity Sheet Quarter 1-MELC 1 Week 1: Respiratory and Circulatory Systems, Working With Other Organ SystemsDocument15 pagesScience Activity Sheet Quarter 1-MELC 1 Week 1: Respiratory and Circulatory Systems, Working With Other Organ SystemsJermil James BaciaNo ratings yet
- Acute Coronary Syndrome: Indra PrasetyaDocument92 pagesAcute Coronary Syndrome: Indra PrasetyaPD-A UB 2017No ratings yet
- CVI PresentationDocument8 pagesCVI PresentationAsad JadoonNo ratings yet
- Vasoactive MedicationsDocument1 pageVasoactive Medicationsapi-635152754No ratings yet
- Lymphatic System Transes 1Document8 pagesLymphatic System Transes 1Sophia Margarette CawalingNo ratings yet
- A A To Raco AbdominalDocument64 pagesA A To Raco AbdominalYgor AlbuquerqueNo ratings yet
- Circulatory System ActivitiesDocument16 pagesCirculatory System ActivitiespatokgayangNo ratings yet
- JAAD CME Aug 2023 - Cutaneous Manifestations of Cardiovascular and Vascular DiseaseDocument39 pagesJAAD CME Aug 2023 - Cutaneous Manifestations of Cardiovascular and Vascular DiseasejchengwangNo ratings yet
- Moyamoya DiseaseDocument12 pagesMoyamoya DiseaseFarida Irawati SiregarNo ratings yet
- RESIT 2014 PAPER: CERVIX, BREAST, THYROID, HEART DISEASE MCQSDocument3 pagesRESIT 2014 PAPER: CERVIX, BREAST, THYROID, HEART DISEASE MCQSDr-Irfan Habib100% (1)
- DefinitionDocument9 pagesDefinitionKhondokar ArafatNo ratings yet
- CARDIO Lab ManualDocument15 pagesCARDIO Lab ManualApril Joy de LimaNo ratings yet
- Cardiovascular Cannulation Products OverviewDocument37 pagesCardiovascular Cannulation Products OverviewAhmadNo ratings yet
- Parts and FunctionsDocument4 pagesParts and FunctionsGerwin SilvestreNo ratings yet
- The circulatory system transports oxygen and nutrientsDocument2 pagesThe circulatory system transports oxygen and nutrientsAmarpreet Kaur100% (1)
- Anesthesia Consideration For Cranipharyngioma Resection in Pediatric PatientDocument5 pagesAnesthesia Consideration For Cranipharyngioma Resection in Pediatric PatientpitriaNo ratings yet
- Nama: Bilqis Inas Nur Hanifah NIM: 021411131041 Obat - Obat Beta BlockersDocument3 pagesNama: Bilqis Inas Nur Hanifah NIM: 021411131041 Obat - Obat Beta BlockersbilqisinasNo ratings yet
- Animal Science (Cardiovascular System)Document23 pagesAnimal Science (Cardiovascular System)Zoyjoy OmpocNo ratings yet
- Hemodynamics For The Bedside Nurse 1CEUDocument7 pagesHemodynamics For The Bedside Nurse 1CEURN333100% (1)
- Circulatory Excretory Systems Mind MapDocument5 pagesCirculatory Excretory Systems Mind MapkiiinNo ratings yet
- Atrial Septal DefectDocument1 pageAtrial Septal DefectChlo14No ratings yet
- Howtotreatrightheart Failure - Tipsforclinicians IneverydaypracticeDocument11 pagesHowtotreatrightheart Failure - Tipsforclinicians Ineverydaypracticefidelurtecho4881No ratings yet
- Venous & Lymphatics, Dr. AngsaDocument40 pagesVenous & Lymphatics, Dr. AngsaKailash KhatriNo ratings yet