You might also like
- Rapid, Reversible Heterolytic Cleavage of Bound HDocument4 pagesRapid, Reversible Heterolytic Cleavage of Bound H陳弘No ratings yet
- Solvent-Dependent Structure of Iridium Dihydride Complexes Different Geometries at Low and High Dielectricity of The MediumDocument10 pagesSolvent-Dependent Structure of Iridium Dihydride Complexes Different Geometries at Low and High Dielectricity of The Mediumxuyijing2007comNo ratings yet
- Effect of Solvents and pH on Electronic Transitions of 2,4-Dihydroxy-5-FluoropyrimidineDocument7 pagesEffect of Solvents and pH on Electronic Transitions of 2,4-Dihydroxy-5-FluoropyrimidinePraveen KumarNo ratings yet
- Introduction to Determining Acidity of HydrocarbonsDocument109 pagesIntroduction to Determining Acidity of HydrocarbonsYoel FriedmanNo ratings yet
- Chemistry Shift 1 Nest 2023Document8 pagesChemistry Shift 1 Nest 2023Hardik JoshiNo ratings yet
- Simulation - ": Gas-Phase Inorganic Chemlstry: Laser Spectroscopy of Calcium and Strontium Monopyrrolate MoleculesDocument4 pagesSimulation - ": Gas-Phase Inorganic Chemlstry: Laser Spectroscopy of Calcium and Strontium Monopyrrolate MoleculesDamxz5No ratings yet
- Gas-Phase Hydroformylation of Propene Over Silica-Supported PPH - Modified Rhodium CatalystsDocument9 pagesGas-Phase Hydroformylation of Propene Over Silica-Supported PPH - Modified Rhodium CatalystsIlireaNo ratings yet
- Biomimetic hydrogen catalystsDocument5 pagesBiomimetic hydrogen catalystsaliNo ratings yet
- Experimental Determination and Thermodynamic Modeling of Methane and Nitrogen Hydrates in The Presence of THF, Propylene Oxide, 1,4-Dioxane and AcetoneDocument12 pagesExperimental Determination and Thermodynamic Modeling of Methane and Nitrogen Hydrates in The Presence of THF, Propylene Oxide, 1,4-Dioxane and AcetoneShurooq TaibNo ratings yet
- 2013 Role of Hydroxyl Radicals During The Competitive EO of Organic Compounds On A BDD AnodeDocument7 pages2013 Role of Hydroxyl Radicals During The Competitive EO of Organic Compounds On A BDD AnodeJosé de Jesús Treviño ReséndezNo ratings yet
- Effect of Ammonia On PT, Ru, RH, and Ni Cathodes During The Alkaline Hydrogen Evolution ReactionDocument13 pagesEffect of Ammonia On PT, Ru, RH, and Ni Cathodes During The Alkaline Hydrogen Evolution ReactionsamypalNo ratings yet
- Thermodynamics of Transfer of Naphthalene and 2-Naphthoic Acid From Water To (Water + Ethanol) Mixtures at T 298:15 KDocument12 pagesThermodynamics of Transfer of Naphthalene and 2-Naphthoic Acid From Water To (Water + Ethanol) Mixtures at T 298:15 KAbdullah MofarrahNo ratings yet
- Molecular Structure: Journal ofDocument8 pagesMolecular Structure: Journal ofYean-baptiste Dupas DecallaisNo ratings yet
- SOLVENT EXCHANGE ON METAL IONSDocument69 pagesSOLVENT EXCHANGE ON METAL IONSgeo angNo ratings yet
- Hydro Borat I OnDocument198 pagesHydro Borat I OnEman MoustafaNo ratings yet
- Catalytic Hydrodeoxygenation Phenols Correlations of KineticDocument9 pagesCatalytic Hydrodeoxygenation Phenols Correlations of KineticNickNo ratings yet
- Organic Chemistry WordDocument19 pagesOrganic Chemistry Wordisnaini safitriNo ratings yet
- 00 B 495395 Ac 3 Cedca 6000000Document28 pages00 B 495395 Ac 3 Cedca 6000000SuhailyShukriNo ratings yet
- The Ionization Constant of Carbonic Acid in Water and The Solubility of Carbon Dioxide in Water and Aqueous Salt Solutions ToDocument8 pagesThe Ionization Constant of Carbonic Acid in Water and The Solubility of Carbon Dioxide in Water and Aqueous Salt Solutions TozibaNo ratings yet
- Keane 1999Document11 pagesKeane 1999sj singhNo ratings yet
- Direct Potentiometric Determination of Fluoride Species by Using Ion-Selective Fluoride ElectrodeDocument10 pagesDirect Potentiometric Determination of Fluoride Species by Using Ion-Selective Fluoride ElectrodeMarielle PerejonNo ratings yet
- A Pulse Radiolysis Study of Coumarin and PDFDocument9 pagesA Pulse Radiolysis Study of Coumarin and PDFSaurav PaulNo ratings yet
- Chapter11 SampleQuestionsDocument16 pagesChapter11 SampleQuestionsHalil EmreNo ratings yet
- Unifying Solution and Surface Electrochemistry: Limitations and Opportunities in Surface ElectrocatalysisDocument7 pagesUnifying Solution and Surface Electrochemistry: Limitations and Opportunities in Surface ElectrocatalysisMiguel Angel SandovalNo ratings yet
- Re-Evaluation of The 2,2-Diphenyl-1-Picrylhydrazyl Free Radical (DPPH) Assay For Antioxidant ActivityDocument10 pagesRe-Evaluation of The 2,2-Diphenyl-1-Picrylhydrazyl Free Radical (DPPH) Assay For Antioxidant Activitycentro surcolombiano de investigación en café uscoNo ratings yet
- Synthesis and Characterization of Metal Complexes of Novel Schiff's Base Ligands Derived From 4-Carboxy Hydrazide-5,6-Diphenyl-3 (2-H) PyridazoneDocument15 pagesSynthesis and Characterization of Metal Complexes of Novel Schiff's Base Ligands Derived From 4-Carboxy Hydrazide-5,6-Diphenyl-3 (2-H) PyridazoneScholedge PublishingNo ratings yet
- The Kinetics of Hydrogen Absorption in Palladium (A - and P-Phase) and Palladium-Silver-AlloysDocument10 pagesThe Kinetics of Hydrogen Absorption in Palladium (A - and P-Phase) and Palladium-Silver-AlloysJose Fernandez AdellNo ratings yet
- Computational and Theoretical Chemistry: SciencedirectDocument7 pagesComputational and Theoretical Chemistry: SciencedirectErwinsyah NurhidayatNo ratings yet
- 3. PAPER - VÍA SAL DE BENCENDIAZONIODocument11 pages3. PAPER - VÍA SAL DE BENCENDIAZONIOValeria Villanueva CervantesNo ratings yet
- 17O NMR Study of Water Exchange On (GD (DTPA) (H2O) ) 2-And (GD (DOTA) (H2O) ) - Related To NMR ImagingDocument8 pages17O NMR Study of Water Exchange On (GD (DTPA) (H2O) ) 2-And (GD (DOTA) (H2O) ) - Related To NMR ImagingGrazi ZaidanNo ratings yet
- BTP - Sem. 7: Topic-Iron Catalyzed CO2 Hydrogenation To Formate Enhanced by Lewis Acid Co-CatalystsDocument13 pagesBTP - Sem. 7: Topic-Iron Catalyzed CO2 Hydrogenation To Formate Enhanced by Lewis Acid Co-CatalystsKUMAR SNEHITNo ratings yet
- Transesterification Kinetics of Phenyl Salicylate 2Document20 pagesTransesterification Kinetics of Phenyl Salicylate 2Lucas de Lima e SousaNo ratings yet
- Rosales 2008Document4 pagesRosales 2008Ezequiel SaavedraNo ratings yet
- Cinetica AA FCDocument4 pagesCinetica AA FCRicardo MartinezNo ratings yet
- NMR Studies of Amnine Species in MEADocument4 pagesNMR Studies of Amnine Species in MEAJohn Jairo RamosNo ratings yet
- Higher-than-Termolecular Proton Transfer in Aqueous Solutions of ImidazoleDocument2 pagesHigher-than-Termolecular Proton Transfer in Aqueous Solutions of ImidazoleUAS TekimNo ratings yet
- Anomaly of The Basicity of Water in Mixed SolventsDocument8 pagesAnomaly of The Basicity of Water in Mixed SolventsRodolfo TravainiNo ratings yet
- Phase Equilibria of Salt-Solvent-CO2 SystemsDocument9 pagesPhase Equilibria of Salt-Solvent-CO2 SystemsJuan Sebastian LopezNo ratings yet
- Polyfunctional Anion Exchanger As Sorbent of Copper (II) and Vanadium (V) IonsDocument4 pagesPolyfunctional Anion Exchanger As Sorbent of Copper (II) and Vanadium (V) IonsKo NwayNo ratings yet
- A Study of The Kinetics and Mechanism of Oxidation of Fluorene by Alkaline Hexacyanoferrate (III)Document7 pagesA Study of The Kinetics and Mechanism of Oxidation of Fluorene by Alkaline Hexacyanoferrate (III)Anonymous eyNieitp2No ratings yet
- Dlelectrlc Properties Electrolyte Solutions. 1. Sodium Iodide in Seven Solvents at Various TemperaturesDocument5 pagesDlelectrlc Properties Electrolyte Solutions. 1. Sodium Iodide in Seven Solvents at Various Temperaturesed roNo ratings yet
- Question BankDocument11 pagesQuestion BankRajeev GuptaNo ratings yet
- ChemistryDocument9 pagesChemistryJoaldo GarciaNo ratings yet
- Kinetics of MethanationDocument12 pagesKinetics of MethanationGabriela Campos DávilaNo ratings yet
- Nucleophilic Substitution and Elimination ReactionsDocument51 pagesNucleophilic Substitution and Elimination ReactionsImam Syafi'iNo ratings yet
- Calculations of The Exchange Current Density For Hydrogen Electrode Reactions PDFDocument6 pagesCalculations of The Exchange Current Density For Hydrogen Electrode Reactions PDFVandam65No ratings yet
- Electrochemistry of Copper in Aqueous Glycine Solutions: Serdar Aksu and Fiona M. DoyleDocument7 pagesElectrochemistry of Copper in Aqueous Glycine Solutions: Serdar Aksu and Fiona M. DoyleRosarioJuyoSalazarNo ratings yet
- pH Measurement FundamentalsDocument6 pagespH Measurement FundamentalsAndré FerrazNo ratings yet
- H. J. Bakker Et Al - Transient Absorption of Vibrationally Excited WaterDocument7 pagesH. J. Bakker Et Al - Transient Absorption of Vibrationally Excited WaterImasmzNo ratings yet
- 9110 Et Et 14Document10 pages9110 Et Et 14ArangaNo ratings yet
- Critical Review of Rate Constant For Reaction of Hydrated ElectronsDocument21 pagesCritical Review of Rate Constant For Reaction of Hydrated ElectronsumairyaqubqaziNo ratings yet
- Enthalpy and hydration of methane hydrateDocument9 pagesEnthalpy and hydration of methane hydrateomeo habibNo ratings yet
- Chlorine 10.1016@0043-13549490118-XDocument11 pagesChlorine 10.1016@0043-13549490118-XSajjad HussainNo ratings yet
- 1 s2.0 S0021951702936201 MainDocument8 pages1 s2.0 S0021951702936201 MainAbdulhamid AliNo ratings yet
- Biophysics 6Document25 pagesBiophysics 6ADITYAROOP PATHAKNo ratings yet
- Chemistry XI - Part-1Document4 pagesChemistry XI - Part-1Janki VernekarNo ratings yet
- Display Article For FreeDocument10 pagesDisplay Article For Free666667No ratings yet
- Reduction Potential - WikipediaDocument28 pagesReduction Potential - WikipediaBabar AliNo ratings yet
- Toluene Vanadium Electrolytic OxidationDocument5 pagesToluene Vanadium Electrolytic Oxidationles_gaidzionis9376No ratings yet
- Ion Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsFrom EverandIon Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsNo ratings yet
- ImidazoleDocument1 pageImidazoleMuhammad JavedNo ratings yet
- M.sc. Inorganic Chemisty Book ListDocument2 pagesM.sc. Inorganic Chemisty Book ListMuhammad JavedNo ratings yet
- Lists of Things Needed For ConferenceDocument1 pageLists of Things Needed For ConferenceMuhammad JavedNo ratings yet
- ExercisesDocument3 pagesExercisesMuhammad JavedNo ratings yet
- GATE Question Parer Chemistry 2007Document21 pagesGATE Question Parer Chemistry 2007Muhammad JavedNo ratings yet
- Nobel Prize List ChemistryDocument24 pagesNobel Prize List ChemistryMuhammad JavedNo ratings yet
- Lists of Things Needed For ConferenceDocument1 pageLists of Things Needed For ConferenceMuhammad JavedNo ratings yet
- Low-Temperature NMR Spectra of Fluoride - Acetic Acid Hydrogen-Bonded Complexes in Aprotic Polar EnvironmentDocument10 pagesLow-Temperature NMR Spectra of Fluoride - Acetic Acid Hydrogen-Bonded Complexes in Aprotic Polar EnvironmentMuhammad JavedNo ratings yet
- Heterocycles Essentials1-2009Document2 pagesHeterocycles Essentials1-2009Aravindan NatarajanNo ratings yet
- Jib 138 2003Document8 pagesJib 138 2003Muhammad JavedNo ratings yet
- AcSIR Advt Jan-2013Document1 pageAcSIR Advt Jan-2013Muhammad JavedNo ratings yet
- Gratz ElDocument12 pagesGratz ElMuhammad JavedNo ratings yet
- Information Brochure OCES/ DGFS - 2013Document13 pagesInformation Brochure OCES/ DGFS - 2013Muhammad JavedNo ratings yet
- Radziszewskis Imidazole SynthesisDocument5 pagesRadziszewskis Imidazole SynthesisZahide Öztaş100% (1)
- GATE 2013 BrochureDocument93 pagesGATE 2013 BrochureSurbhi KumariNo ratings yet
- Cy 10Document24 pagesCy 10Joy KunduNo ratings yet
- AcSIR Advt Jan-2013Document1 pageAcSIR Advt Jan-2013Muhammad JavedNo ratings yet
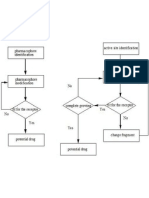
- Flow Charts of Two Strategies of Structure Based Drug DesignDocument1 pageFlow Charts of Two Strategies of Structure Based Drug DesignMuhammad JavedNo ratings yet
- API DrugsDocument1 pageAPI DrugsMuhammad JavedNo ratings yet