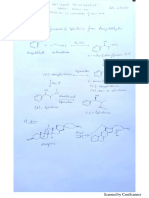
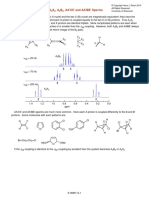
You might also like
- Cheat SheetDocument5 pagesCheat Sheetkittenface92% (13)
- Daily Assessment RecordDocument4 pagesDaily Assessment Recordapi-342236522100% (2)
- SHORT STORY Manhood by John Wain - Lindsay RossouwDocument17 pagesSHORT STORY Manhood by John Wain - Lindsay RossouwPrincess67% (3)
- CH3 OxudationDocument12 pagesCH3 OxudationWaqasNo ratings yet
- Complete Guide To E-Commerce TechnologyDocument420 pagesComplete Guide To E-Commerce Technologypilatus100% (4)
- Photocatalytic Decolorization of Bismarck Brown RDocument170 pagesPhotocatalytic Decolorization of Bismarck Brown Rgeorge.ejaam8975No ratings yet
- Critical Review of Rate Constant For Reaction of Hydrated ElectronsDocument21 pagesCritical Review of Rate Constant For Reaction of Hydrated ElectronsumairyaqubqaziNo ratings yet
- Selected Redox ReactionsDocument5 pagesSelected Redox ReactionsWilfredo LlanaNo ratings yet
- Environmental Photochemistry: Is Iron Oxide (Hematite) An Active Photocatalyst? A Comparative Study: A-Fe20,, Zno, Ti02Document9 pagesEnvironmental Photochemistry: Is Iron Oxide (Hematite) An Active Photocatalyst? A Comparative Study: A-Fe20,, Zno, Ti02Kristanto WahyudiNo ratings yet
- 1 s2.0 S0021951702936201 MainDocument8 pages1 s2.0 S0021951702936201 MainAbdulhamid AliNo ratings yet
- 1 s2.0 0160412079900187 MainDocument6 pages1 s2.0 0160412079900187 MainSaurav BhattacharjeeNo ratings yet
- Gopalan 1994Document7 pagesGopalan 1994artinels9No ratings yet
- Oxidation Reduction ReactionsDocument40 pagesOxidation Reduction ReactionsLock SeriesNo ratings yet
- Theoretical Estimation of The Apparent Rate Constants For Ozone Decomposition in Gas and Aqueous Phases Using Ab Initio CalculationsDocument7 pagesTheoretical Estimation of The Apparent Rate Constants For Ozone Decomposition in Gas and Aqueous Phases Using Ab Initio CalculationsMarkus MeierNo ratings yet
- Permanganate Oxidation Mechanisms of Alkylarenes PDFDocument13 pagesPermanganate Oxidation Mechanisms of Alkylarenes PDFJuanda BarbosaNo ratings yet
- 1D Shebaldina2004Document5 pages1D Shebaldina2004Akshayan RNo ratings yet
- Adsorption of Co Ni Cu and ZN On Amorphous Hydrous MnO2 From 1-1 Electrplyte Solutions-LibreDocument11 pagesAdsorption of Co Ni Cu and ZN On Amorphous Hydrous MnO2 From 1-1 Electrplyte Solutions-LibreNaeem AkramNo ratings yet
- Palladium-Catalyzed Cross-CouplingDocument27 pagesPalladium-Catalyzed Cross-CouplingAntonio Cortez D Lara XDNo ratings yet
- Oxidation and ReductionDocument2 pagesOxidation and ReductionClaudine VelascoNo ratings yet
- Pertemuan 10-11 ch05 - Oxidation Reduction ReactionsDocument40 pagesPertemuan 10-11 ch05 - Oxidation Reduction ReactionsArifah YumnaNo ratings yet
- Department of Chemistry, Ben-Gurion University of The Negev, Beer-Sheva, Israel Department of Biological Chemistry, The College of Judea and Samaria, Ariel, IsraelDocument19 pagesDepartment of Chemistry, Ben-Gurion University of The Negev, Beer-Sheva, Israel Department of Biological Chemistry, The College of Judea and Samaria, Ariel, IsraelRafael Ricardo Celin ManceraNo ratings yet
- Hemsheela Model School Durgapur Term-Ii Examination-2022 Chemistry Marking Scheme Class-XiDocument6 pagesHemsheela Model School Durgapur Term-Ii Examination-2022 Chemistry Marking Scheme Class-XiBaichitra MondalNo ratings yet
- Permanganate Oxidation Mechanisms of Alkylarenes: Mukul ChauhanDocument12 pagesPermanganate Oxidation Mechanisms of Alkylarenes: Mukul ChauhanGio De La Rosa HernandezNo ratings yet
- Znfeldh Chen 2012Document7 pagesZnfeldh Chen 2012Baka NataNo ratings yet
- Grade 12 Chemistry Handout for Electrochemistry LessonsDocument30 pagesGrade 12 Chemistry Handout for Electrochemistry LessonsHagre Tube100% (1)
- Effects of Synthesis Conditions On The Structural and Electrochemical PropertiesDocument5 pagesEffects of Synthesis Conditions On The Structural and Electrochemical PropertiesjoseNo ratings yet
- ElectrochemistryFinal Rev 2020 LECDocument104 pagesElectrochemistryFinal Rev 2020 LECsimonjohn spanglerNo ratings yet
- Applied Catalysis B: Environmental: O. Shmychkova, T. Luk'yanenko, A. Yakubenko, R. Amadelli, A. VelichenkoDocument6 pagesApplied Catalysis B: Environmental: O. Shmychkova, T. Luk'yanenko, A. Yakubenko, R. Amadelli, A. VelichenkoPeter KozlikhinNo ratings yet
- Nucleophilic Substitution Questions - PKBDocument12 pagesNucleophilic Substitution Questions - PKBPawan BabelNo ratings yet
- Micro Reactor For Chlorophenols OxidationDocument9 pagesMicro Reactor For Chlorophenols OxidationBANUNo ratings yet
- Desulphurisation of Lead Paste PDFDocument6 pagesDesulphurisation of Lead Paste PDFAswathi IndustriesNo ratings yet
- Chem DA PDFDocument8 pagesChem DA PDFvarsh kollaNo ratings yet
- Accepted Manuscript: 10.1016/j.jallcom.2017.09.306Document22 pagesAccepted Manuscript: 10.1016/j.jallcom.2017.09.306Bautista SanderNo ratings yet
- Determination of Hardness in Water by Complexometric TitrationDocument52 pagesDetermination of Hardness in Water by Complexometric TitrationNamrata BajpaiNo ratings yet
- Eu 3+ doped Ba 2 LaV 3 O 11 phosphors for warm white LEDsDocument28 pagesEu 3+ doped Ba 2 LaV 3 O 11 phosphors for warm white LEDssivaji naikNo ratings yet
- Electrode Determination: Ionization Constants and HydrolysisDocument14 pagesElectrode Determination: Ionization Constants and HydrolysisHanyszShalNo ratings yet
- Removal of Phosphate From Aqueous Solutions Using Calcined Metal Hydroxides Sludge Waste Generated From ElectrocoagulationDocument8 pagesRemoval of Phosphate From Aqueous Solutions Using Calcined Metal Hydroxides Sludge Waste Generated From ElectrocoagulationLamine tech & scienceNo ratings yet
- Study On Production of Free Hydroxyl Radical and Its Reaction With Salicylic Acid at Lead Dioxide ElectrodeDocument7 pagesStudy On Production of Free Hydroxyl Radical and Its Reaction With Salicylic Acid at Lead Dioxide ElectrodePeter KozlikhinNo ratings yet
- OxidationDocument16 pagesOxidationCoralsimmerNo ratings yet
- Reserach Article 2Document15 pagesReserach Article 2Umesh ChandraNo ratings yet
- Micellar Effects Upon Oxidation of Organic Sulfides by Anionic OxidantsDocument9 pagesMicellar Effects Upon Oxidation of Organic Sulfides by Anionic Oxidantssalem emhamedNo ratings yet
- 2013 Role of Hydroxyl Radicals During The Competitive EO of Organic Compounds On A BDD AnodeDocument7 pages2013 Role of Hydroxyl Radicals During The Competitive EO of Organic Compounds On A BDD AnodeJosé de Jesús Treviño ReséndezNo ratings yet
- Introdution To Physical Chemistry Third Edition - 2Document53 pagesIntrodution To Physical Chemistry Third Edition - 2FarhadNo ratings yet
- A Review On The Generation of Superoxide IonDocument15 pagesA Review On The Generation of Superoxide IonDr. Balmukund yadavNo ratings yet
- Lead Dioxide 5Document11 pagesLead Dioxide 5Khobaib HayatNo ratings yet
- ReviewDocument9 pagesReviewUseless MeNo ratings yet
- Pateli Et Al. 2020 Electrochemical Oxidation in DES PreprintDocument26 pagesPateli Et Al. 2020 Electrochemical Oxidation in DES PreprintEkRA GoRaYANo ratings yet
- CFC-xyz: X, Number of C-1 Y, Number of H+1 Z, Number of F CFC-115: CF3CF2Cl (Low K Due To The Low Polarizability of F Long Half Time in Troposphere)Document55 pagesCFC-xyz: X, Number of C-1 Y, Number of H+1 Z, Number of F CFC-115: CF3CF2Cl (Low K Due To The Low Polarizability of F Long Half Time in Troposphere)Haoyu ZhaoNo ratings yet
- Low-Temperature NMR Spectra of Fluoride - Acetic Acid Hydrogen-Bonded Complexes in Aprotic Polar EnvironmentDocument10 pagesLow-Temperature NMR Spectra of Fluoride - Acetic Acid Hydrogen-Bonded Complexes in Aprotic Polar EnvironmentMuhammad JavedNo ratings yet
- 2000 Effects of Oxidant and Solvent On The Liquid Phase Cyclohexane Oxidation Catalyzed by Ce Exchanged Zeolite YDocument7 pages2000 Effects of Oxidant and Solvent On The Liquid Phase Cyclohexane Oxidation Catalyzed by Ce Exchanged Zeolite YYash GokaniNo ratings yet
- Solutions To Preparatory Problems: Problem 1. Graphite OxideDocument25 pagesSolutions To Preparatory Problems: Problem 1. Graphite OxideNebojsaZecNo ratings yet
- Instrumental Analysis Techniques: Chemiluminescence and BioluminescenceDocument30 pagesInstrumental Analysis Techniques: Chemiluminescence and BioluminescenceSozdar ArgoshiNo ratings yet
- Chemistry ExDocument12 pagesChemistry ExAmit KingNo ratings yet
- Oxidation of tert-butanethiol with air using Mo-containing hydrotalcite-like compounds and their derived mixed oxides as catalystsDocument18 pagesOxidation of tert-butanethiol with air using Mo-containing hydrotalcite-like compounds and their derived mixed oxides as catalystsElisabeta StamateNo ratings yet
- Mercury FulminateDocument11 pagesMercury FulminateAnoshän IndreswaränNo ratings yet
- Sdarticle 010Document67 pagesSdarticle 010geo angNo ratings yet
- Chapter 11 - The Chemistry of Ethers, Epoxides, Glycols, and SulfidesDocument7 pagesChapter 11 - The Chemistry of Ethers, Epoxides, Glycols, and SulfidesJohn SmithNo ratings yet
- Oxidation Reduction ReactionsDocument33 pagesOxidation Reduction ReactionsSaeful Ghofar100% (2)
- Cation ExcluderDocument3 pagesCation ExcluderAnand RajNo ratings yet
- Fourth International Conference on Non-Aqueous Solutions: Vienna 1974From EverandFourth International Conference on Non-Aqueous Solutions: Vienna 1974V. GutmannNo ratings yet
- Practice Makes Perfect in Chemistry: Oxidation-ReductionFrom EverandPractice Makes Perfect in Chemistry: Oxidation-ReductionRating: 5 out of 5 stars5/5 (1)
- 25th International Congress of Pure and Applied Chemistry: Plenary Lectures Presented at the 25th International Congress of Pure and Applied Chemistry, Jerusalem, Israel 6–11 July 1975From Everand25th International Congress of Pure and Applied Chemistry: Plenary Lectures Presented at the 25th International Congress of Pure and Applied Chemistry, Jerusalem, Israel 6–11 July 1975Rating: 5 out of 5 stars5/5 (1)
- Nakanishi 2004Document9 pagesNakanishi 2004Saurav PaulNo ratings yet
- High Mobility Hole TransportDocument7 pagesHigh Mobility Hole TransportSaurav PaulNo ratings yet
- Optical studies reveal nature of "blue phaseDocument2 pagesOptical studies reveal nature of "blue phaseSaurav PaulNo ratings yet
- Voltagedependent Optical Activity of A Twisted Nematic Liquid CrystalDocument3 pagesVoltagedependent Optical Activity of A Twisted Nematic Liquid CrystalSaurav PaulNo ratings yet
- Dielectric Materials: Dielectric Materials Are and Used Principally in andDocument7 pagesDielectric Materials: Dielectric Materials Are and Used Principally in andSaurav PaulNo ratings yet
- Scanned by CamscannerDocument2 pagesScanned by CamscannerSaurav PaulNo ratings yet
- CV 1st PartDocument3 pagesCV 1st PartSaurav PaulNo ratings yet
- 7-Ion CHDocument3 pages7-Ion CHSaurav PaulNo ratings yet
- A Highly Selective and Sensitive Fluorescent Chemosensor For Fe in Physiological Aqueous SolutionDocument2 pagesA Highly Selective and Sensitive Fluorescent Chemosensor For Fe in Physiological Aqueous SolutionSaurav PaulNo ratings yet
- Tetrahedron Letters: Hiromasa Mitsudera, Chao-Jun LiDocument3 pagesTetrahedron Letters: Hiromasa Mitsudera, Chao-Jun LiSaurav PaulNo ratings yet
- Designing A Green Synthesis: of StartingDocument3 pagesDesigning A Green Synthesis: of StartingSaurav PaulNo ratings yet
- Firth BMJ1991 Chaos Predicting UnpredictableDocument4 pagesFirth BMJ1991 Chaos Predicting UnpredictableRakesh Babu100% (1)
- Structure Problem Solving Using H and C NMR Spectral Data Tutorial SessionDocument12 pagesStructure Problem Solving Using H and C NMR Spectral Data Tutorial SessionSaurav PaulNo ratings yet
- SINTETIS Ibuprofen PDFDocument6 pagesSINTETIS Ibuprofen PDFRodolfo Angulo OlaisNo ratings yet
- CL O CL: 4. Green Chemistry in Day-to-Day LifeDocument2 pagesCL O CL: 4. Green Chemistry in Day-to-Day LifeSaurav PaulNo ratings yet
- Ahluwalia 2004Document1 pageAhluwalia 2004Saurav PaulNo ratings yet
- Ahluwalia 2004Document4 pagesAhluwalia 2004Saurav PaulNo ratings yet
- Application of IR Spectroscopy and Interpretation of IR SpectrumDocument10 pagesApplication of IR Spectroscopy and Interpretation of IR SpectrumMuhammad HussnainNo ratings yet
- Cross Dehydrogenative CouplingDocument10 pagesCross Dehydrogenative CouplingAnonymous rm2rf6No ratings yet
- 1455787242CHE P12 M30 EtextDocument8 pages1455787242CHE P12 M30 EtextSaurav PaulNo ratings yet
- 1455787242CHE P12 M30 EtextDocument8 pages1455787242CHE P12 M30 EtextSaurav PaulNo ratings yet
- Factors Affecting Vibrational Frequenciesand IR Spectroscopy of HydrocarbonsDocument17 pagesFactors Affecting Vibrational Frequenciesand IR Spectroscopy of HydrocarbonsMuhammad HussnainNo ratings yet
- Applications of 13C NMR in Structural DeterminationDocument13 pagesApplications of 13C NMR in Structural DeterminationSaurav PaulNo ratings yet
- 1455787138CHE P12 M29 EtextDocument7 pages1455787138CHE P12 M29 EtextSaurav PaulNo ratings yet
- 13C NMR ConceptsDocument15 pages13C NMR ConceptsSaurav PaulNo ratings yet
- 1455787432CHE P12 M33 E-TextDocument12 pages1455787432CHE P12 M33 E-TextSaurav PaulNo ratings yet
- 1455786764CHE P12 M24 E-TextDocument10 pages1455786764CHE P12 M24 E-TextSaurav PaulNo ratings yet
- Account Statement From 1 Sep 2019 To 11 Aug 2020: TXN Date Value Date Description Ref No./Cheque No. Debit Credit BalanceDocument2 pagesAccount Statement From 1 Sep 2019 To 11 Aug 2020: TXN Date Value Date Description Ref No./Cheque No. Debit Credit BalanceSaurav PaulNo ratings yet
- AA'BB' SpectraDocument11 pagesAA'BB' SpectraSaurav PaulNo ratings yet
- 1455877785CHE P12 M35 EtextDocument8 pages1455877785CHE P12 M35 EtextSaurav PaulNo ratings yet
- Best Home Oxygen Concentrators-Lowest Prices & Fast Shipping (Oxygen Machines) - 2021 - YuwellDocument1 pageBest Home Oxygen Concentrators-Lowest Prices & Fast Shipping (Oxygen Machines) - 2021 - YuwellPelayanan ResusitasiNo ratings yet
- U-KLEEN Moly Graph MsdsDocument2 pagesU-KLEEN Moly Graph MsdsShivanand MalachapureNo ratings yet
- InteliLite AMF20-25Document2 pagesInteliLite AMF20-25albertooliveira100% (2)
- Prof. Ed - Assessment and Evaluation of Learning Part 1-4Document8 pagesProf. Ed - Assessment and Evaluation of Learning Part 1-4Marisol Altobar PalatinoNo ratings yet
- Technology Consulting: Amruta Kulkarni Anu Abraham Rajat JainDocument9 pagesTechnology Consulting: Amruta Kulkarni Anu Abraham Rajat JainRajat JainNo ratings yet
- Openness To Experience: Intellect & Openness: Lecture Notes 8Document8 pagesOpenness To Experience: Intellect & Openness: Lecture Notes 8Danilo Pesic100% (1)
- Oas Community College: Republic of The Philippines Commission On Higher Education Oas, AlbayDocument22 pagesOas Community College: Republic of The Philippines Commission On Higher Education Oas, AlbayJaycel NepalNo ratings yet
- Body Shaming Among School Going AdolesceDocument5 pagesBody Shaming Among School Going AdolesceClara Widya Mulya MNo ratings yet
- 2 Druid StreetDocument4 pages2 Druid StreetthamestunnelNo ratings yet
- Project Report On AdidasDocument33 pagesProject Report On Adidassanyam73% (37)
- Service Positioning and DesignDocument3 pagesService Positioning and DesignSaurabh SinhaNo ratings yet
- Solutions: Spheres, Cones and CylindersDocument13 pagesSolutions: Spheres, Cones and CylindersKeri-ann MillarNo ratings yet
- Grace Lipsini1 2 3Document4 pagesGrace Lipsini1 2 3api-548923370No ratings yet
- Corporate Governance in SMEsDocument18 pagesCorporate Governance in SMEsSana DjaanineNo ratings yet
- Serv7107 V05N01 TXT7Document32 pagesServ7107 V05N01 TXT7azry_alqadry100% (6)
- Business EnvironmentDocument12 pagesBusiness EnvironmentAbhinav GuptaNo ratings yet
- 2nd QUARTER EXAMINATION IN P. E 2019-2020Document3 pages2nd QUARTER EXAMINATION IN P. E 2019-2020Lyzl Mahinay Ejercito MontealtoNo ratings yet
- Advanced Presentation Skills: Creating Effective Presentations with Visuals, Simplicity and ClarityDocument15 pagesAdvanced Presentation Skills: Creating Effective Presentations with Visuals, Simplicity and ClarityGilbert TamayoNo ratings yet
- spl400 Stereo Power Amplifier ManualDocument4 pagesspl400 Stereo Power Amplifier ManualRichter SiegfriedNo ratings yet
- Eportfile 4Document6 pagesEportfile 4api-353164003No ratings yet
- 20ME901 Automobile Engineering Unit 3Document74 pages20ME901 Automobile Engineering Unit 36044 sriramNo ratings yet
- Strategic Flexibility: The Evolving Paradigm of Strategic ManagementDocument3 pagesStrategic Flexibility: The Evolving Paradigm of Strategic Managementnanthini kanasanNo ratings yet
- Abstract Photo CompositionDocument2 pagesAbstract Photo Compositionapi-260853196No ratings yet
- IPO Fact Sheet - Accordia Golf Trust 140723Document4 pagesIPO Fact Sheet - Accordia Golf Trust 140723Invest StockNo ratings yet
- ElectricalDocument30 pagesElectricalketerNo ratings yet
- 4 TheEulerianFunctions - 000 PDFDocument16 pages4 TheEulerianFunctions - 000 PDFShorouk Al- IssaNo ratings yet
- Answer Questions Only: Chemical Engineering DeptDocument2 pagesAnswer Questions Only: Chemical Engineering DepthusseinNo ratings yet