Professional Documents
Culture Documents
SDM Biophysics 09062008
Uploaded by
alex_sopilniakOriginal Description:
Original Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
SDM Biophysics 09062008
Uploaded by
alex_sopilniakCopyright:
Available Formats
Index
1. An Introduction to Photosynthesis and Its Applications 2
1.1 The Basics. ............................................................................................................................ 2
1.2 Reaction Centers and Antennae. ........................................................................................... 3
1.3 Photosynthetic Electron Transfer. ......................................................................................... 5
1.4 Carbon Fixation. ................................................................................................................... 7
1.5 Increasing CO
2
levels. ........................................................................................................... 9
1.6 Photosynthesis and respiration. ............................................................................................. 9
1.7 Diversity of Photosynthetic Organisms. ............................................................................. 10
1.8 Evolution. ............................................................................................................................ 10
1.9 Early Events. ....................................................................................................................... 11
1.10 Structure Determinations. ................................................................................................. 11
1.11 Similarities Between Reaction Centers. ............................................................................ 12
1.12 Implications of Photosynthesis Studies. ........................................................................... 12
1.13 Rapid electron transfer reactions. ..................................................................................... 13
1.14 Organic molecules mimicking reaction centers. ............................................................... 13
1.15 Genetic modification and protein engineering. ................................................................. 14
1.16 Genetic modification of higher plants............................................................................... 15
1.17 Future Directions. ............................................................................................................. 15
2. The Structure and Function of BioMolecules. .......................................................................... 17
2.1. The Structure of Molecules. ............................................................................................... 17
2.1.1 The Born-Oppenheimer Approximation ...................................................................... 17
2.1.2 Finding the approximate wavefunctions for H
2
+
. ........................................................ 18
2.1.3 Bonding and antibonding molecular orbitals ............................................................... 19
2.1.4 The structure of diatomic molecules. ........................................................................... 20
2.1.5 The structure of heteronuclear diatomic molecules ..................................................... 24
2.1.6 The variation principle ................................................................................................. 24
2.1.7 Hybridization and The Structure of Polyatomic Molecules. ....................................... 28
2.1.8 Hckel Molecular Orbital Theory ................................................................................ 35
2.1.9 Symmetry ..................................................................................................................... 41
2.1.10 Quantum Chemistry ................................................................................................... 42
Configuration Interaction, Hartree-Fock, DFT ............................................................................. 42
2.2 Non-Covalent Interactions. ................................................................................................. 42
2.2.1 Charge-Charge and dipole-dipole interactions. ........................................................... 42
2.2.2 Van der Waals forces ................................................................................................... 44
2.2.3. The intermolecular potential energy ........................................................................... 48
2.2.4. The hydrogen bond ..................................................................................................... 49
2.2.5 The hydrophobic interaction ........................................................................................ 52
4. Macromolecules and Biomolecules .......................................................................................... 56
4.1 Structure and Dynamics. ..................................................................................................... 56
4.1.1 The different levels of structure. .................................................................................. 56
4.2 Random coils ...................................................................................................................... 59
4.3 The Structure of Proteins .................................................................................................... 63
4.3.1 The amino acids. .......................................................................................................... 63
4.3.2 The electronic structure of the peptide bond. ............................................................... 65
4.3.3 The total energy of a protein and its energy landscape ................................................ 67
4.3.4 The ohelix and the |-sheet. ....................................................................................... 71
4.4 Protein Folding.................................................................................................................... 78
4.5 Respiration 85
4.6 Membranes .......................................................................................................................... 85
4.6.1 Formation of the lipid bilayer ...................................................................................... 90
4.6.2 Transport across a membrane ...................................................................................... 91
5. The photosynthetic pigments .................................................................................................... 95
5.2 Spectroscopic Properties of Chlorophylls ........................................................................... 99
5.3 Carotenoids ....................................................................................................................... 103
6. BioMolecules Viewed with Electronic Spectroscopy ............................................................. 106
6.1 Absorption......................................................................................................................... 106
6.1.1 The transition dipole moment. ................................................................................... 107
6.1.2 The Einstein Coefficients ........................................................................................... 110
6.1.3 Lambert-Beers Law .................................................................................................. 112
6.1.4 Franck-Condon factors .............................................................................................. 115
6.1.5 The electronic spectra of polyatomic molecules ........................................................ 119
6.1.6 Linear Dichroism ....................................................................................................... 121
6.1.7. The photochemistry of vision ................................................................................... 127
6.1.8 Linewidths and lineshapes ......................................................................................... 130
6.2 The Fates of the Electronic Excited States ....................................................................... 143
6.2.1 Fluorescence and Phosphorescence. .......................................................................... 143
7. The Light-Harvesting Photosynthetic Antenna ...................................................................... 148
7.1 General concept ................................................................................................................ 148
7.2. A bit of history ................................................................................................................. 149
7.3. Why antennas? ................................................................................................................. 149
7.4. Classes of antennas .......................................................................................................... 149
7.5. Physical Principles of Antenna Function ......................................................................... 152
7.6 How to measure antenna function..................................................................................... 157
8. Excitons and Excitation Energy Transfer. .............................................................................. 163
8.1. Introduction ...................................................................................................................... 163
8.2. The Excitonically Coupled Dimer ................................................................................... 163
8.2.1 The exciton states ....................................................................................................... 163
8.2.2 The redistribution of oscillator strength ..................................................................... 166
8.2.3 The coupling matrix element ..................................................................................... 167
8.5 The disordered dimer ........................................................................................................ 168
8.6 Energy transfer within the dimer. ..................................................................................... 169
8.7 The Frster Equation......................................................................................................... 170
8.8 Hopping vs The Disordered Exciton Model and Redfield Relaxation ............................. 173
9. Reaction Centers ..................................................................................................................... 175
9.1.The Structure of the Photosynthetic Reaction Center. ...................................................... 175
9.2. Primary Electron Transfer Reactions in Bacterial Photosynthetic Reaction Centers ...... 182
9.3. Biological Electron Transfer: The Marcus model ........................................................... 186
- 1 -
BIOPHYSICS
The Structure of Biological Matter and Photosynthesis
Biological molecules, proteins, DNA, membranes are at the basis of life. How precisely they do
this is unknown, but it is certain that our understanding of life and life and life processes goes
hand in hand with our understanding of the fundamental properties of biomolecules, either as
single molecules, or in interaction with other molecules or ultimately in the living cell or in a
living organism. Biomolecules are per definition complex, even a small biomolecule will
contain a few thousand atoms, or at least 10
4
electrons and even the simplest biomolecule, like
the simple protein myoglobin, can occur in an almost infinite number of configurations.
Although it is not impossible to construct a Hamiltonian for such a system, including its
interaction with the environment is difficult (if not impossible) and solving its Schrdinger
equation is again another story. Consequently all our understanding of biomolecules is based on
approximations. This lecture series is meant to acquaint students with the properties of living
matter. It is also meant to show that when you go from the level of atoms to the level of
molecules and then to the level of biomolecules some properties of the lower level survive, while
others become completely irrelevant. Furthermore new physical properties appear and often in a
non-intuitive way. It is the goal of (bio-)physics to find out which properties survive and which
emerge when we try to understand the hydrogen atom of life, the protein. In this lecture series
we will concentrate on the photosynthetic proteins: antenna proteins that harvest the solar light,
reaction centers that drive an ultrafast charge separation, electron transfer proteins, and how
these proteins function in the intact photosynthetic membrane.
Fig. 1 Cartoon of the photosynthetic membrane. The photosynthetic membrane is a lipid bilayer which
contains the photosynthetic proteins. The photosynthetic pigment proteins, Photosystem 1 and Photosystem 2
(PS1 & PS2) are multi-protein complexes in which a few hundred chlorophylls and other pigments are bound.
Light is absorbed by a light-harvesting antenna (about 200 Chls per PS1 or PS2), the electronic excitation
energy is transferred to either PS1 or PS2 where a trans-membrane charge separation is initiated. PS2
oxidizes H
2
O to generate electrons, protons and produce O
2
, PSI reduces NADP
+
. The electrons are
transported from water via PSII to PSI and pass the cyt b
6
f complex, where the available free energy is used to
transport additional H
+
across the membrane to establish a transmembrane ApH. The pH-gradient is used by
the ATP-synthase to produce ATP.
- 2 -
1. An Introduction to Photosynthesis and Its Applications
1.1 The Basics.
Sunlight plays a much larger role in our sustenance than we may expect: all the food we eat and
all the fossil fuel we use is a product of photosynthesis, which is the process that converts energy
in sunlight to chemical forms of energy that can be used by biological systems. Photosynthesis is
carried out by many different organisms, ranging from plants to bacteria (Figure 1.1). The best
known form of photosynthesis is the one carried out by higher plants and algae, as well as by
cyanobacteria and their relatives, which are responsible for a major part of photosynthesis in
oceans. All these organisms convert CO
2
(carbon dioxide) to organic material by reducing this
gas to carbohydrates in a rather complex set of reactions. Electrons for this reduction reaction
ultimately come from water, which is then converted to oxygen and protons. Energy for this
process is provided by light, which is absorbed by pigments (primarily chlorophylls and
carotenoids). Chlorophylls absorb blue and red light and carotenoids absorb blue-green light
(Figure 1.2), but green and yellow light are not effectively absorbed by photosynthetic pigments
in plants; therefore, light of these colors is either reflected by leaves or passes through the leaves.
This is how plants are green. Why is another story!
Figure 1.1. Examples of photosynthetic
organisms: leaves from higher plants flanked
by colonies of photosynthetic purple bacteria
(left) and cyanobacteria (right).
- 3 -
Figure 1.2. Absorption spectrum of isolated chlorophyll and
carotenoid species. The color associated with the various
wavelengths is indicated above the graph.
Other photosynthetic organisms, such as cyanobacteria and red algae, have additional pigments
called phycobilins that are red or blue and that absorb the colors of visible light that are not
effectively absorbed by chlorophyll and carotenoids. Yet other organisms, such as the purple and
green bacteria (which, by the way, look fairly brown under many growth conditions), contain
bacteriochlorophyll that absorbs in the near-infrared in addition to in the blue part of the
spectrum. These bacteria do not evolve oxygen, but perform photosynthesis under anaerobic
(oxygen-less) conditions. These bacteria efficiently use near-infrared light for photosynthesis.
(Near-)Infrared is light with wavelengths above 700 nm that cannot be seen by the human eye;
some bacterial species can use infrared light with wavelengths of up to 1000 nm. However, most
pigments are not very effective in absorbing ultraviolet light (<400 nm), which also cannot be
seen by the human eye. Light with wavelengths below 330 nm becomes increasingly damaging
to cells, but virtually all light at these short wavelengths is filtered out by the atmosphere (most
prominently the ozone layer) before reaching the earth. Even though most plants are capable of
producing compounds that absorb ultraviolet light, an increased exposure to light around 300 nm
has detrimental effects on plant productivity.
1.2 Reaction Centers and Antennae.
Photosynthetic pigments come in a huge variety: there are many different types of
(bacterio)chlorophyll, carotenoids, and phycobilins, differing from each other in their precise
chemical structure. Pigments generally are bound to proteins, which provide the pigment
molecules with the appropriate orientation and positioning with respect to each other. Light
energy is absorbed by individual pigments or by groups of pigments, but is not used immediately
by these pigments for energy conversion. Instead, the light energy is rapidly (i.e. rapid on the
timescale of the natural lifetime of a chlorophyll excited state: a few nanoseconds) transferred to
chlorophylls that are in a special protein environment where the actual energy conversion event
occurs: the light energy is used to transfer an electron to a neighboring pigment (see fig. 1.3).
- 4 -
Pigments and protein involved with this actual primary electron transfer event together are called
the reaction center. A large number of pigment molecules (100-5000), collectively referred to as
antenna, "harvest" light and transfer the light energy to the same reaction center. The purpose is
to maintain a high rate of electron transfer in the reaction center, even at lower light intensities.
Fig. 1.3 Basic concept of antenna function. Light is absorbed by a pigment (carotenoid,
chlorophyll) and the electronic excited state is transferred on an ultrafast timescale from
pigment to pigment until chlorophylls in the reaction center become excited. In this reaction
center the excited chlorophyll transfers an electron to a nearby electron acceptor and in this way
the energy is stored.
Many antenna pigments transfer their light energy to a single reaction center by having this
energy "hop" to another antenna pigment, and yet to another, etc., until the energy is "trapped" in
the reaction center. Each step of this energy transfer must be very efficient to avoid a large loss
in the overall transfer process, and the association of the various pigments with proteins ensures
that transfer efficiencies are high by having appropriate pigments close to each other, and by
having an appropriate molecular geometry of the pigments with respect to each other. An
exception to the rule of protein-bound pigments are green bacteria with very large antenna
systems: a large part of these antenna systems consists of a "bag" (named chlorosome) of up to
several thousand bacteriochlorophyll molecules that interact with each other and that are not in
direct contact with protein.
In many systems the size of the photosynthetic antenna is flexible, and photosynthetic organisms
growing at low light (in the shade, for example) generally will have a larger number of antenna
pigments per reaction center than those growing at higher light intensity. However, at high light
intensities (for example, in full sunlight) the amount of light that is absorbed by plants exceeds
the capacity of electron transfer initiated by reaction centers. Plants have developed means to
convert some of the absorbed light energy to heat rather than to use the absorbed light
necessarily for photosynthesis. However, in particular the first part of photosynthetic electron
transfer in plants is rather sensitive to overly high rates of electron transfer, and part of the
photosynthetic electron transport chain may be shut down when the light intensity is too high;
this phenomenon is known as photoinhibition.
- 5 -
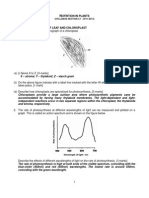
1.3 Photosynthetic Electron Transfer.
The initial electron transfer (charge separation) reaction in the photosynthetic reaction center sets
into motion a long series of redox (reduction-oxidation) reactions, passing the electron along a
chain of cofactors and filling up the "electron hole" on the chlorophyll, much like in a bucket
brigade. All photosynthetic organisms that produce oxygen have two types of reaction centers,
named photosystem II and photosystem I (PS II and PS I, for short), both of which are
pigment/protein complexes that are located in specialized membranes called thylakoids. In
eukaryotes (plants and algae), these thylakoids are located in chloroplasts (organelles in plant
cells) and often are found in membrane stacks (grana) (Figures 1.4 and 1.5). Prokaryotes
(bacteria) do not have chloroplasts or other organelles, and photosynthetic pigment-protein
complexes either are in the membrane around the cytoplasm or in invaginations thereof (as is
found, for example, in purple bacteria), or are in thylakoid membranes that form much more
complex structures within the cell (as is the case for most cyanobacteria) (Figure 1.6).
Figure 1.4. Artist's rendition of a leaf (bottom),
thylakoids within a chloroplast (middle), and a
photosystem in thylakoids (top).
- 6 -
Figure 1.5. Electron micrograph of a thin
section of an algal cell. The cup-shaped
structure around the edge of the cell (open near
the top) is the chloroplast. The structures
resembling mostly parallel lines in the
chloroplast are the thylakoid membranes.
Courtesy of Dr. Ken Hoober.
- 7 -
Figure 1.6. Freeze-fracture electron micrograph
of a cyanobacterial cell, showing exposed
thylakoid membrane surfaces (upper right).
Thylakoids are stacked like folded pancakes,
and this image represents a surface-cut through
these thylakoids.
All chlorophyll in oxygenic organisms is located in thylakoids, and is associated with PS II, PS I,
or with antenna proteins feeding energy into these photosystems. PS II is the complex where
water splitting and oxygen evolution occurs. Upon oxidation of the reaction center chlorophyll in
PS II, an electron is pulled from a nearby amino acid (tyrosine) which is part of the surrounding
protein, which in turn gets an electron from the water-splitting complex. From the PS II reaction
center, electrons flow to free electron carrying molecules (plastoquinone) in the thylakoid
membrane, and from there to another membrane-protein complex, the cytochrome b
6
f complex.
The other photosystem, PS I, also catalyzes light-induced charge separation in a fashion basically
similar to PS II: light is harvested by an antenna, and light energy is transferred to a reaction
center chlorophyll, where light-induced charge separation is initiated. However, in PS I electrons
are transferred eventually to NADP (nicotinamide adenosine dinucleotide phosphate), the
reduced form of which can be used for carbon fixation. The oxidized reaction center chlorophyll
eventually receives another electron from the cytochrome b
6
f complex. Therefore, electron
transfer through PS II and PS I results in water oxidation (producing oxygen) and NADP
reduction, with the energy for this process provided by light (2 quanta for each electron
transported through the whole chain).
Figure 1.7: Overview of photosynthetic processes as they occur in plants, algae, and
cyanobacteria.
Note that to oxidize water a very oxidizing potential is required, while to reduce NADP+,
requires a very reducing potential, implying that, although the two photosystems are built from
the same compounds, protein and chlorophyll, the potential at which they operate must be very
different. In fact the oxidation potential generated by photosystem II is so oxidizing that with a
- 8 -
relatively high probability the system is damaged (something unwanted becomes oxidized) and
the PSII reaction center must be repaired. The system is taken apart, the damaged unit is replaced
and the reaction center is reassembled. In a plant in normal light every PSII reaction center must
be repaired every half hour!!
A schematic overview of the light capture and charge transfer processes is provided in Figure
1.7.
1.4 Carbon Fixation.
Electron flow from water to NADP requires light and is coupled to generation of a proton
gradient across the thylakoid membrane. This proton gradient is used for synthesis of ATP
(adenosine triphosphate), a high-energy molecule. ATP and reduced NADP that resulted from
the light reactions are used for CO
2
fixation in a process that is independent of light. CO
2
fixation
involves a number of reactions that is referred to as the Calvin-Benson cycle. The initial CO
2
fixation reaction involves the enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase
(RuBisCO), which can react with either oxygen (leading to a process named photorespiration and
not resulting in carbon fixation) or with CO
2
. The probability with which RuBisCO reacts with
oxygen vs. with CO
2
depends on the relative concentrations of the two compounds at the site of
the reaction. In all organisms CO
2
is by far the preferred substrate, but as the CO
2
concentration
is very much lower than the oxygen concentration, photorespiration does occur at significant
levels. To boost the local CO
2
concentration and to minimize the oxygen tension, some plants
(referred to as C
4
plants) have set aside some cells within a leaf (named bundle-sheath cells) to
be involved primarily in CO
2
fixation, and others (named mesophyll cells) to specialize in the
light reactions: ATP, CO
2
and reduced NADP in mesophyll cells is used for synthesis of 4-
carbon organic acids (such as malate), which are transported to bundle sheath cells. Here the
organic acids are converted releasing CO
2
and reduced NADP, which are used for carbon
fixation. The resulting 3-carbon acid is returned to the mesophyll cells. The bundle sheath cells
generally do not have PS II activity, in order to minimize the local oxygen concentration.
However, they retain PS I, presumably to aid in ATP synthesis. Even though C4 plants have
reduced amounts of photorespiration, the amount of ATP they need per amount of CO
2
fixed is a
little higher than in other plants, and therefore their total production rate is similar to that of
plants with higher rates of photorespiration.
Some plants living in desert climates, such as cacti, keep their stomates closed during the day to
minimize evaporation (stomates are openings in the leaf surface to enhance gas exchange). These
plants take up CO
2
during the night when the stomates are open, and temporarily bind the CO
2
to
organic acids in the leaf. During the day the CO
2
is released from the acids and used for
photosynthesis. Plants using this mechanism of CO
2
fixation are called CAM (Crassulacean Acid
Metabolism) plants (Figure 1.8).
- 9 -
Figure 1.8. The light reactions of
photosynthesis stop when the sun goes down.
However, CO
2
fixation can continue as long as
ATP and NADPH is available. In cacti and
other succulents CO
2
uptake by the plant occurs
primarily at night.
1.5 Increasing CO
2
levels.
The amount of overall CO
2
fixation in plants growing under optimal conditions is limited
primarily by the amount of CO
2
available. Therefore, the increase of CO
2
in the atmosphere will
lead to somewhat higher rates of plant growth in environments where the CO
2
concentration
limits growth rates. This is usually the case in an agricultural setting, where nutrients and water
availability are not limiting. However, also in natural conditions, where limitations other than the
CO
2
concentration will generally limit plant productivity, plant productivity has been found to
often increase upon increasing the CO
2
concentration.
1.6 Photosynthesis and respiration.
Virtually all oxygen in the atmosphere is thought to have been generated through the process of
photosynthesis. Obviously, all respiring organisms (including plants) utilize this oxygen and
produce CO
2
. Thus, photosynthesis and respiration are interlinked, with each process depending
on the products of the other. The global amount of photosynthesis is on the order of a trillion kg
of dry organic matter produced per day, and respiratory processes convert about the same
amount of organic matter to CO
2
. A large part (probably the majority) of photosynthetic
productivity occurs in open oceans, mostly by oxygenic prokaryotes. Without photosynthesis, the
oxygen in the atmosphere would be depleted within several thousand years. It should be
emphasized that plants respire just like any other higher organism, and that during the day this
respiration is masked by a higher rate of photosynthesis.
- 10 -
1.7 Diversity of Photosynthetic Organisms.
Even though plants are the most visible representatives of photosynthetic organisms, it should be
emphasized that many other types of photosynthetic organisms exist. All photosynthetic bacteria
other than the cyanobacteria and their relatives use only one photosystem, and for
thermodynamic reasons they cannot use water as the ultimate electron donor. Instead, they can
use reduced compounds such as H
2
S as donor. However, CO
2
fixation occurs in these organisms.
Some of these photosynthetic bacteria appear to have retained an evolutionary ancient
arrangement of their photosynthetic apparatus, and are of interest for the analysis of evolutionary
relationships of photosynthetic systems.
An extensive group of these photosynthetic bacteria, the heliobacteria, was discovered rather
recently in the 1980s. The first representative of this group was isolated by the group of Dr.
Howard Gest from a soil sample collected on the campus of Indiana University, and this isolation
was the result of a fortunate coincidence of serendipitous events. Analysis of the heliobacterial
reaction center has helped to lay the basis for the current concept that all photosynthetic reaction
centers from the large variety of photosynthetic organisms are related to each other. The majority
of bacteria cannot be maintained in pure culture (that is, without other organisms). This has
essentially limited analysis of photosynthetic prokaryotes to the relatively small group of
organisms that can be grown in pure culture. It is likely that the actual diversity of photosynthetic
organisms is much larger than is known thus far. Indeed, species with novel photosynthetic
properties are reported virtually every year. For example, recently an organism was reported that
has chlorophyll d (a chlorophyll that is very rare in nature) as the main pigment. Moreover,
several years ago, previously undetected and very small chlorophyll a/b-containing prokaryotes
were recognized to be the major contributors to photosynthetic production in the open ocean.
This emphasizes that much relating to biodiversity and photosynthesis is still to be discovered,
and that these discoveries are not limited to tropical rainforests and other ecological settings of
large popular interest.
1.8 Evolution.
In eukaryotes, photosynthesis takes place in the chloroplast, which has long been known to have
prokaryotic (bacterial) features. Chloroplasts are thought to have evolved from a
cyanobacterium (or close relative) that was in a symbiotic relationship with a eukaryotic, non-
photosynthetic cell and was engulfed inside this cell. The cyanobacterium and the eukaryotic cell
presumably were in a mutually beneficial relationship (endosymbiosis), with the photosynthetic
organism sharing some of its produced carbohydrates with the host, and the host providing the
photosynthetic bacterium with other compounds. The prokaryote slowly gave up its
independence as well as its cell wall, and some of its genetic information was transferred to the
nucleus of its eukaryotic host. The resulting chloroplast maintains a small, prokaryote-like
circular DNA of its own (DNA is material carrying genetic information); this DNA contains the
genetic blueprint to make many of the membrane proteins needed in the chloroplast, which
apparently are not easily targeted to and/or transported into the chloroplast. Occasionally,
photosynthetic organisms are found where the chloroplast has retained a little more of the
original cyanobacterial features. For example, in algae such as Cyanophora paradoxa plastids
(called cyanelles) are found that resemble cyanobacteria in their overall morphology as well as in
the fact that they are surrounded by a cell wall.
- 11 -
Not all chloroplasts have resulted from a single endosymbiotic event, but apparently from
multiple events that occurred independently. Chloroplasts from higher plants and many green
algae probably all result from the same endosymbiotic event, whereas chloroplasts from red and
brown algae and from diatoms are the result of one or more other events. The situation is even
more complicated in cryptomonads, a type of algae, and chlorachniophytes, photosynthetic
amoebae, which apparently are the result of an endosymbiotic event of an eukaryotic alga in an
eukaryotic host. The nucleus of the endosymbiont has been mostly degraded, resulting in a
chloroplast enveloped by four membranes.
1.9 Early Events.
Chlorophyll is used by all photosynthetic organisms as the link between excitation energy
transfer and electron transfer. Of particular note is the rate with which these transfer reactions
need to occur. As the lifetime of the excited state is only several nanoseconds (1 nanosecond (ns)
is 10
-9
s), after absorption of a quantum, energy transfer and charge separation in the reaction
center must have occurred within this time period. Energy transfer rates between pigments are
very rapid, and charge separation in reaction centers occurs in 3-30 picoseconds (1 picosecond
(ps) is 10
-12
s). Subsequent electron transfer steps are significantly slower (200 ps - 20 ms) but,
nonetheless, the electron transport chain is sufficiently fast that at least a significant part of the
absorbed sunlight can be used for photosynthesis. However, in the presence of excess light,
damage may occur, which may originate from the formation of chlorophyll in a "triplet state". In
a triplet state two electrons in the outer shell have identical rather than opposite spin orientation.
This triplet chlorophyll readily reacts with oxygen, leading to the very reactive singlet oxygen,
which can damage proteins. To counter this damaging reaction, carotenoids are usually present
in close vicinity to chlorophylls. Many carotenoids efficiently "quench" triplet states of
chlorophyll, thus avoiding formation of singlet oxygen. Chlorophyll in its free form is very toxic
in the light in the presence of oxygen, because a close interaction with carotenoids is not always
available under such circumstances. Therefore, all chlorophyll in a cell in aerobic organisms is
bound to proteins, generally with carotenoids bound to the same protein.
1.10 Structure Determinations.
Because of the strict requirements of positioning of pigments and electron transfer intermediates
to allow efficient energy and electron transfer and minimal damage, the structure of pigment-
protein complexes involved in photosynthesis is critical. With the exception of specific antenna
complexes (such as phycobilisomes in cyanobacteria and chlorosomes in green bacteria),
pigment-binding proteins are usually hydrophobic membrane proteins. This initially hampered
attempts to elucidate the structure of these complexes, as membrane proteins do not readily form
the well-ordered crystals that are needed for high-resolution X-ray diffraction studies. However,
in the 1980s the first structure of a membrane protein complex, the photosynthetic reaction
center from a purple bacterium, was determined at high resolution (about 3 ; in comparison, the
distance between neighboring atoms in a molecule is about 1 ). Investigators from the Max
Planck Institute in Martinsried (Germany) who were involved with this work, most notably
Hartmut Michel and Johann Deisenhofer, received a Nobel Prize in chemistry for this research.
Since then, the structure of various reaction centers and antenna complexes has been determined
at resolutions between 2 and 4 . Figure 1.9 presents the structure of the photosynthetic reaction
center from the purple bacterium Rhodobacter sphaeroides. The mechanism of operation of this
complex will be discussed extensively below.
- 12 -
Figure 1.9. Molecular structure of a bacterial
reaction center. Cofactors are indicated in red. The
three proteins making up the reaction center are in
blue, yellow, and green. Ribbons in the protein
represent helices through the membrane. Wire-like
protein regions represent domains outside of the
membrane.
1.11 Similarities Between Reaction Centers.
Surprisingly, structural comparison of reaction centers from different photosynthetic systems
showed that these reaction centers are basically similar to each other in terms of their overall
three-dimensional structure. The basic reaction center unit consists of a protein complex with 10
transmembrane helices originating from either two identical protein subunits or from two similar
polypeptides of common evolutionary origin. Each of these polypeptides contribute five
membrane-spanning helices, and bind 2-3 chlorophylls (or, in the case of anoxygenic bacteria,
bacteriochlorophylls). The fourth membrane spanning helices from each subunit are held
together by two chlorophylls, which are the chlorophylls in the reaction center that can be
oxidized (give up an electron) upon excitation. Directly associated with the reaction center are
proteins that bind antenna pigments. In the case of PS I and similarly organized reaction centers
from green and heliobacteria, the antenna portion, with six transmembrane helices, is covalently
attached to the N-terminal end of the reaction center proteins.
1.12 Implications of Photosynthesis Studies.
Photosynthesis has been studied in significant detail and photosynthetic systems are used
frequently for development and application of advanced technologies, because photosynthetic
- 13 -
systems are fairly well understood, are complex, and often undergo rather unusual biochemical
reactions. Some examples are provided below.
1.13 Rapid electron transfer reactions.
A major difficulty in measuring enzyme kinetics at relatively short time scales (less than 1 ms) is
that "traditional" enzyme reactions require a mixing of substrate and enzyme, which usually
takes a relatively long time. Kinetic analysis of light-driven reactions such as photosynthetic
electron transport have a great advantage in this respect: reactions can be triggered simply by a
light pulse, which can be even shorter than 1 ps. Moreover, many of the components
participating in electron transfer have different absorption spectra depending on whether they are
in the oxidized or reduced form. Using laser (and other) spectroscopy methods it is possible to
follow the electron around on a timescale between 1 ps and several ms. The primary charge
separation occurs in several ps, and reactions become gradually slower as they involve
components that are further away from the reaction center. Because of the fast speed of early
reactions, the electron and the "electron hole" are physically separated rapidly by a large distance
(the electron generally has traveled about 2-3 nm to the other side of the membrane within 1 ns
after charge separation), so that back reactions (charge recombinations) are not favorable
anymore. Unpaired electrons on reactants that are transiently formed during redox reactions
involving transfer of a single electron in many instances can be detected using electron
paramagnetic resonance (EPR) and derived techniques (including ENDOR, electron nuclear
double resonance, and ESEEM, electron spin echo envelope modulation). Many of these
techniques can be used to kinetically follow redox reactions, and provide detailed information
regarding electron spin distributions etc. Therefore, photosynthetic membranes and reaction
centers have a prominent place as experimental systems in biochemistry and biophysics.
1.14 Organic molecules mimicking reaction centers.
Reaction centers are essentially an assembly of cofactors, held in the appropriate position and
orientation by the protein environment. Several groups have used the natural system as a model
to design organic molecules where the equivalents of the different cofactors are linked together
by covalent bonds of various lengths. The result is the creation of a number of sophisticated
molecules that serve as "artificial reaction centers". The more advanced molecules consist of two
chlorophyll-type molecules linked together (one serves as the electron donor, the other as the
acceptor), with the electron-accepting molecule linked to two quinones, which serve as electron
acceptors in the natural system. The electron donating chlorophyll analog is linked covalently to
a carotenoid, which can donate an electron to the oxidized chlorophyll. Upon excitation of the
chlorophyll, a charge separation occurs resulting in an oxidized carotenoid and a reduced
quinone. This charge-separated state is formed with high efficiency. An example of such a
molecule is presented in Figure 1.10.
- 14 -
Figure 1.10. Molecular model of an artificial reaction center. The
two bulky structures in the middle are chlorophyll-like
components, flanked by a carotenoid (left) and quinones (right).
Courtesy of Dr. Devens Gust.
Such molecules can be introduced into liposomes (artificial membrane vesicles) in a specific
orientation, and when these are excited by light, a charge separation will occur across the
liposome membrane. This results in an electric potential or proton gradient across the liposome
membrane, which may be used for a variety of purposes, including ATP synthesis (the latter
requires introduction of the ATP synthesizing enzyme into the liposome membrane). The groups
of Ana and Tom Moore and Devens Gust at Arizona State University are leaders in
developments in this area.
1.15 Genetic modification and protein engineering.
Because of the ease of detailed functional analysis of reactions and their rates in photosynthetic
systems, reaction center complexes are frequently used to determine the consequences of small
alterations in the polypeptides on the functional characteristics of the cofactors. Changes at
single amino acid residues in the reaction center complex are sufficient to introduce large
changes in the properties of cofactors, which in turn leads to altered electron transfer rates and
efficiencies. Single amino acid changes at specific sites in the protein are easily introduced by
genetic modification techniques, and resulting functional changes can be studied. An elegant
example of such an approach is the modification of the midpoint redox potential of the
bacteriochlorophyll in the reaction center of purple bacteria. The midpoint redox potential is
correlated with the ease with which an electron is given off after excitation and is taken up by the
oxidized bacteriochlorophyll. For instance creating or deleting hydrogen bonds between the
protein and the bacteriochlorophyll changed the midpoint redox potential of this
bacteriochlorophyll in a rather predictable manner. In this way, reaction center complexes can be
built with different oxidizing strengths, and effects on reaction rates and ultimately the
effectiveness of alternate electron donors can be determined. Mutational analysis of
photosynthesis proteins is simple in several bacterial systems. The reasons why this is so in
selected cyanobacteria and purple and green bacteria are that (1) foreign DNA is taken up by the
cell spontaneously or is introduced easily by other means such as electroporation ("electric
shock"), (2) once the DNA is inside it is incorporated into the organism's genome at one
predictable and specific site by means of a process named homologous double recombination,
and (3) the organism can be propagated without relying on photosynthesis, for example using an
added carbohydrate source.
Genetic approaches involving directed mutagenesis as described above have proven to be very
useful in studying photosynthetic electron transfer and will be of increasing relevance for the
design of photosynthetic organisms for biotechnological uses (see below). By this method the
function of a large number of genes has been probed, and the role of individual domains and
residues has been determined. Genomic sequencing projects are very useful in this respect, and
the complete DNA sequence of one photosynthetic organism is already known. From the DNA
sequence, the potential of the organism can be determined. The entire 3,573,470 nucleotide-long
genomic DNA sequence of the transformable cyanobacterium Synechocystis 6803 was
determined by Satoshi Tabata and coworkers at the Kazusa DNA Research Institute in Japan.
- 15 -
This organism is used by several researchers to elucidate the role of many proteins thought to be
involved in photosynthetic or other physiological processes. Meanwhile, other groups are
working on the genomic sequence of two purple bacteria. With the genomic sequence in hand,
the role of specific genes can be found by amplifying the gene of interest by means of
polymerase chain reaction, cloning it into a plasmid, replacing the gene by a selectable marker
(i.e., a piece of DNA coding for a protein inactivating a particular antibiotic, thus conferring
antibiotic resistance), and analyzing the functional characteristics of resulting mutants. A
website, CyanoBase (http://www.kazusa.or.jp/cyano/cyano.orig.html), has been established to
facilitate searching of the genomic sequence of Synechocystis 6803, and a related site,
CyanoMutants (http://www.kazusa.or.jp/cyano/mutants/), has been developed that
accommodates information regarding targeted mutations and their effects in this organism.
1.16 Genetic modification of higher plants.
Globally, improving plant productivity by genetic means has been an important goal for many
years. Even though initially it was hoped that crop productivity could be boosted by "improving"
the photosynthesis process, it has become increasingly apparent that improved productivity will
be best accomplished by genetic modification of crop plants to introduce insect or pathogen
resistance, or to yield improved vitality under marginal conditions (for example, at high salinity,
which has become a significant factor in many agricultural areas because of continued
irrigation). A pertinent example is the development of varieties of cotton and other crops that
express a pro-toxin from a bacterium, Bacillus thuringiensis, that is converted to a toxin in the
mid gut of particular insects such as caterpillars but not in other life forms. This allows efficient
biological control as long as the insects do not develop resistance to this compound.
Genetic modifications to intrinsically and significantly improve the photosynthesis process have
not yet been successful. The reason for the apparent inability to "improve" the photosynthesis
process itself presumably is related to the fact that photosynthetic systems have evolved over a
relatively long period of time, and that the selection factors have not changed significantly in
recent history. This has led to the emergence of a very effective photosynthetic apparatus that is
difficult to improve upon by simply changing some amino acid residues or by introducing or
deleting some genes. If relatively simple changes could have significantly improved the
photosynthesis process per se, Mother Nature would have already found these as natural
mutations are rather frequent. However, it is possible that significant progress in this area can
made in the future, if new design paradigms for enzyme function and specificity can be
developed. For example, if protein structures (particularly the structure of the active site) can be
better modeled and predicted, one should be able to further improve upon the RuBisCO
specificity of CO
2
vs. oxygen. It is also important in this respect to determine what the rate-
limiting step in the process is under natural conditions. Even though more effective light capture
by crop plants might be considered by introduction of antenna pigments that absorb in the green
and yellow region of the light spectrum, light capture and the light reactions usually are not
limiting plant productivity in agricultural settings. Therefore, such modifications in a plant will
result in increased productivity only in light-limiting settings.
1.17 Future Directions.
Light energy is cheap, clean, and essentially inexhaustible. With limited supplies of fossil fuel
and increasing concern about CO
2
emissions, further development of technologies that make use
- 16 -
of solar energy is inevitable. Current silicon-based technologies for the harvesting of solar
energy require a very energy-intensive production process and even though they have improved
significantly over the years in their efficiency, further development of photosynthesis-based
technologies for energy collection is certainly warranted. Today, in the USA and in Europe
major initiatives are in progress to make the connection between the natural process of
photosynthesis and the production of a fourth-generation solar cell that will produce a solar
fuel.
However, photosynthesis and related processes can be applied to many more areas than just
solar energy conversion. Realizing that novel designs and applications of light-mediated
processes have enormous promise in the next decade and beyond, Arizona State University has
started an initiative, Project Ingenhousz, named after the 18th century physician who discovered
that light is needed for oxygen evolution by plants, and that only green parts of the plant carry
out this process. The goal of the initiative is to capitalize on research progress and ideas in this
area, and to more effectively interface academia, where many of the discoveries are made, with
the private sector, where such discoveries are worked out further and applied.
For example, there are a myriad of possible applications of artificial reaction centers and related
molecules in nanotechnology. Many synthetic pigments also have found biomedical uses in
tumor detection, as they -for unknown reasons- tend to accumulate preferentially in tumors and
are highly fluorescent and thus easily detectable in a patient whom is being operated on to
surgically remove a tumor.
In the biotechnology field, photosynthetic organisms are likely to play an increasing role in
(over)production of enzymes, pharmaceuticals, nutraceuticals, etc., which until now are
produced primarily by genetically modified heterotrophic microorganisms such as yeast and
selected bacteria. A major advantage of photosynthetic organisms is that no fixed-carbon source
needs to be added for growth and, therefore, production costs are lower and the chances of
contamination with other microorganisms are less. There are several ways to modify organisms
to have them (over)produce useful compounds. One is to introduce specific genes under a strong
promoter, leading to high expression of these genes and to synthesis of a "new" enzyme. Another
way is to delete genes so that substrates will accumulate. For this to be optimally successful the
metabolic pathways of an organism need to be fairly well understood, and genomic DNA
sequences are an important step in this direction. A third way to produce a new compound is to
utilize an existing enzyme and to modify the site where the substance to be converted (the
substrate) binds, so that a different substrate can be bound and a different product can be formed.
With any of these approaches, selection for randomly generated combinatorial mutants with
specific properties also has been proven to be effective. With an increasing arsenal of genomic
sequences, and with improving knowledge regarding determinants of protein structure and
cofactor binding, any of these three approaches are very promising.
Another potential application of photosynthetic organisms is in bioremediation. Bioremediation
is the clean-up of environmental (soil or water) pollutants by biological means. An example is
the biological breakdown of toxic organic compounds into innocuous products. Also,
remediation of nitrate from drinking water supplies is becoming an increasingly pressing issue.
The advantage of using photosynthetic organisms is that no external energy source needs to be
- 17 -
provided for growth of the organism if it is in the light, making these organisms very suitable for
remediation of aqueous surface environments.
Another utilization of photosynthetic organisms is to have these organisms use solar energy to
produce clean-burning fuels. Even under natural conditions some photosynthetic systems such as
algae can produce hydrogen, which probably is the cleanest fuel as it reacts with oxygen to
produce water. However, the cheapest and most universal electron donor of all, water, upon
oxidation in PS II forms oxygen, which is not very safe in combination with hydrogen. For this
reason methods have been sought to temporally separate oxygen evolution and hydrogen
production in algae; in the laboratory this can be achieved by sulfur deprivation, which
preferentially inhibits photosynthesis. Another option would be to use photosynthetic organisms
for methane production. Even though methane upon combustion will form CO
2
, the overall
atmospheric CO
2
balance would not be disturbed as an equal amount of CO
2
will have been
taken out of the atmosphere upon methane production by the photosynthetic organism.
Research in photosynthesis in all its facets has proven to have opened many doors in a variety of
disciplines, ranging from biophysics to plant physiology. Progress has been driven by an
interdisciplinary approach to this complex, yet fascinating, spectrum of problems, challenges,
and opportunities. Photosynthesis is the basis of our food and energy supply, and innovative
utilization of solar energy is likely to be of increasingly critical importance in the future. This,
together with novel uses of photosynthetic principles for other purposes, make it likely that
photosynthesis and its applications will help to shape an increasingly broad area of exciting
discoveries and innovative ideas.
2. The Structure and Function of BioMolecules.
Biomolecules like proteins and DNA are molecules. They derive their structure from the same
fundamental principles as very simple molecules like H
2
+
or O
2
or C
2
H
4
. However, proteins may
easily consist of a chain of100 covalently linked amino acids, since such a chain has a lot of
conformational freedom, to fold it into the conformation with the minimum energy requires
involves a large number of weak interactions (Van der Waals forces, electrostatic, H-bonds).
Also the properties of the surrounding solvent (often water with salt) play a crucial role. In this
chapter we will review the essential physics of molecules and then discuss the weak interactions
that in the end determine the shape and function of biomolecules
2.1. The Structure of Molecules.
Since even the smallest molecule consists of three particles (2 nuclei, one electron) solving its
Schrodinger equation requires approximations. The most fundamental approximation in
molecular physics is the Born-Oppenheimer approximation.
2.1.1 The Born-Oppenheimer Approximation
The Born-Oppenheimer assumes that the nuclei are static on the timescale of electronic motion.
It allows us to write the total molecular wavefunction as a product of an electronic and a nuclear
wavefunction, where the latter is only dependent on the nuclear coordinates.
( ) ( ) ( )
B A n e B A
R R R r R R r , . , , ,
1 1
= (2.1.1)
- 18 -
where ( ) R r
e
,
1
is the wavefunction for the electron and ( )
B A n
R R , the wavefunction for the
nuclei. Solving the Schrodinger eq. with eq. 2.1.1 yields the electronic energy (plus the nuclear
repulsion term), which acts as a potential energy for the nuclear motion. A typical potential
energy curve as a function of the nuclear configuration) is shown in fig 2.1.1. In first order this
is approximated by a parabolic function and the nuclear wavefunctions are given by those of the
harmonic oscillator.
E
e
R
Fig. 1.2 Electronic energy as a function of internuclear distance R
D
e
R
eq
2.1.2 Finding the approximate wavefunctions for H
2
+
.
In its most simple form one can describe the ground state of H
2
+
by a linear combination of the
two1s orbitals located on each of the two H atoms:
B A MO
s s 1 1 + ~ + (2.1.2)
It is not difficult to see that this is indeed a bonding molecular (because it spreads over the whole
molecule) orbital.
Fig 2.1.1 illustrates an example of an electronic energy potential curve. A stable molecule
is found at an internuclear distance R
eq
. The nuclei move in this potential. Ignoring
translation and rotation for the moment, one observes that around the minimum, the
potential for the nuclei is (almost) harmonic and consequently one can describe the
nuclear motion by that of a harmonic oscillator.
- 19 -
2.1.3 Bonding and antibonding molecular orbitals
It is not difficult to see that
B A MO
s s 1 1 ~ + (2.1.3)
represents an anti-bonding molecular orbital
Fig. 2.1.3. The molecular
potential energy curve for H
2
+
.
The upper curve is discussed
below). The equilibrium bond
length corresponds to the energy
minimum.
Fig. 2.1.2. (a) The orbital overlap responsible for the bonding in H
2
+
and (b) the constructive
interference in the internuclear region. (c) The orbital amplitude in a plane containing the two
nuclei, and (d) the corresponding electron density. (b) is a sideview of (c).
- 20 -
2.1.4 The structure of diatomic molecules.
(a) The hydrogen molecule
Fig 2.1.4. (a) The orbital overlap responsible for antibonding in H
2
+
and (b) the
destructive interference in the internuclear region. (c) The orbital amplitude in a
plane containing the two nuclei, and (d) the corresponding electron density (b) is a
side view of (c).
Fig. 2.1.5. A molecular orbital energy level
diagram for orbitals constructed from (1s,
1s)-overlap, the separation of the levels
corresponding to the equilibrium bond
length.
Fig. 2.1.6. The ground electronic
configuration of H
2
is obtained by
accommodating the two electrons in the
lowest available orbital (the bonding orbital.
- 21 -
(b) Molecular orbital description of the hydrogen molecule: electron-electron
interaction
( ) ( ) { } ( ) ( ) { } ( ) ( ) { } ( ) ( ) ( ) ( ) { } 1 2 2 1
2
1
2 2 1 1 ) 1 2 1 2 , 1
2
1
2
| o | o
|
.
|
\
|
+ + + = b a b a S
H
(2.1.4a)
( )( ) ( )
)
`
+ + + V + V =
R r r r r r
e m H
b a b a
e
1 1 1 1 1 1
4 2
12 2 2 1 1
0
2 2
2
2
1
2
tc
(2.1.4b)
One can use the H
2
wavefunction to calculate the energies of the bonding and anti-bonding
energies E
b
and E
ab
, using elliptical coordinates:
Fig.2.1.7. The ground electronic
configuration of the four-electron molecule
He
2
has two bonding electrons and two
antibonding electrons. It has a higher energy
than the separated atoms, and so He
2
is
unstable.
A
B
u
r
A
r
B
Fig. 2.1.8 Elliptical coordinates:
u =
s u s u
s s
=
s s
+
=
d d d R d
R
r r
R
r r
B A
B A
v v t
t
v v
) (
8
1
2 0 ;
1 1 ;
1 ;
2 2 3
- 22 -
(c) o- and t-molecular orbitals
Fig. 2.1.9. (a) the constructive interference
leading to the formation of a 2po-bonding
orbital and (b) the corresponding
antibonding MO.
Fig. 2.1.10. (a) The interference
of 2p
x
- or 2p
y
-AOs leading to the
formation of a 2pt-bonding
orbital and (b) the corresponding
antibonding orbital. Note that for
the t-orbitals the contribution to
the binding energy of a molecule
is relatively small
- 23 -
(d) s,p-overlap
Fig. 2.1.11. The molecular
orbital energy level
diagram for (2p, 2p)-
overlap. While simple
overlap considerations
suggest the order in (a),
the order often found in
practice is that shown in
fig 2.1.12
Fig. 2.1.12 Variation of the o- and t-orbital energies of Period 2 homonuclear
diatomics.
- 24 -
A measure of the extent to which two orbitals overlap is the overlap integral S
B A B A
d S + + = + + =
}
t * (2.1.5)
.
2.1.5 The structure of heteronuclear diatomic molecules
A heteronuclear diatomic molecule is a molecule of the form AB; chemically interesting species
include CO and HCl. The principal consequence of the presence of different atoms is that the
electron distribution in the bond is no longer symmetrical between the atoms because it may be
energetically more favourable for the charge to drift towards one of the atoms. The range of bond
types, from nonpolar through polar to ionic is captured in MO theory by writing the LCAO as:
B B A A MO
c c + + + = + (2.1.6)
where
A
+ and
B
+ are the AOs contributing to the MO and c
A
and c
B
are the coefficients. The
proportion of
A
+ in the bond is
2
A
c , and the proportion of
B
+ is
2
B
c . A nonpolar bond has
2 2
B A
c c = and a pure ionic bond has one coefficient zero (so that A
+
B
-
would have 0 =
A
c and
1 =
B
c ). A polar bond would have unequal, nonzero coefficients.
2.1.6 The variation principle
The question, of course is how to find the values of the coefficients, and relate them to
the energy of the orbital. The key is the variation principle, which states: I f an arbitrary
wavefunction is used to calculate the energy, then the value obtained is never less than the
true energy. The arbitrary wavefunction is called the trial function. The principle implies that if
we guess the form of an MO (e.g. guess values of the coefficients in eq. (3.1.6) and calculate the
energy of an electron that occupies this MO, then we never get an energy lower than the true
energy. Moreover, the trial MO with the lowest energy is the best of that kind that we can
Fig. 2.1.13. Overlapping s- and p-orbitals. (a)
End-on overlap leads to non-zero overlap and
to the formation of an axially symmetric o-
bond. (b) Broad-side overlap leads to no net
accumulation of electron density in the
internuclear region.
- 25 -
construct. Of course, we might get a lower energy if we use a more complicated wavefunction
(for example by taking a linear combination of several AOs on each atom), but of that specific
kind we have constructed the optimum MO.
We need a systematic way of finding the optimum coefficients in an LCAO. The method
can be illustrated by using the minimal basis set for an MO; this is a basis set that is the simplest
possible for constructing an LCAO. For instance, in the case of HCl it would consist of a
hydrogen 1s-orbital (H1s) and a chlorine 3p-orbital (Cl3p). In general, the minimal basis set for
an AB molecules is ( )
B A
+ + , , and the trial wavefunction is the LCAO given in eq. (2.1.6). This
trial function is not normalized as it stands, because the coefficients can take arbitrary values,
and so in the following we can not assume that 1
2
= +
}
t d .
The energy of an electron described by a trial wavefunction is the expectation value of
the hamiltonian H. For a non-normalized wavefunction this is:
t t d d H E + + + + =
} }
* *
(2.1.7)
From now on we consider only real wavefunctions (not essential, just lazy). We now search for
values of the coefficients in the trial function that minimize the value of E. This is a standard
problem in calculus: we seek the coefficients
B A
c c , such that:
0 = c c
A
c E and 0 = c c
B
c E (2.1.8)
The first step is to express the two integrals in terms of the coefficients. The denominator is:
( )( )
( ) t t t t
t t
d c c d c d c d c c c c
d c c c c d
B A B A B B A A B A B A B B A A
B B A A B B A A
+ + + + + + = + + + + + + =
+ + + + + + = +
} } } }
} }
2 2
2 2 2 2 2 2 2 2
2
The individual AOs are normalized, and so the first two integrals are equal to 1. The third
integral is the overlap integral S, eq. (2.1.5). Therefore the denominator is
S c c c c d
B A B A
2
2 2 2
+ + = +
}
t . The numerator is
( ) ( )
( ) t
t t
d H c c H c H c
d c c H c c d H
B A B A B B B A A A
B B A A B B A A
}
} }
+ + + + + + + +
= + + + + + + = + +
2
2 2
There are some complicated integrals in this expression, but we can denote them by constants.
t o d H
A A A
}
+ + = , t o d H
B B B
}
+ + = , t | d H
B A
}
+ + = (2.1.9)
Then
B A B B A A
c c c c d H | o o t 2
2 2
+ + = + +
}
(2.1.10)
A
o , which is negative (see below), is called the Colulomb integral and can be interpreted as the
energy of an electron when it is in the orbital
A
+ on atom A with nucleus B close by (and
- 26 -
likewise for
B
o . In a homonuclear diatomic molecule
B A
o o = . | is called the resonance
integral (for classical reasons), and vanishes when the orbitals do not overlap; at equilibrium
bond lengths it is normally negative. We can therefore suspect that the strength of the bond is
directly determined by the size of | . The complete expression for E is
S c c c c
c c c c
E
B A B A
B A B B A A
2
2
2 2
2 2
+ +
+ +
=
| o o
(2.1.11)
The minimum is found by differentiation with respect to the two coefficients and using eqs.
(2.1.11). The result is:
( ) ( )
( ) ( ) 0
0
= +
= +
B B A
B A A
c E c ES
c ES c E
o |
| o
(2.1.12)
These are called the secular equations. They have a solution if the determinant of the
coefficients, the secular determinant vanishes; that is, if
0 =
E ES
ES E
B
A
o |
| o
(2.1.13)
This determinant expands to a quadratic equation in E, which may be solved. Its two roots give
the energies of the bonding and antibonding MOs formed from the basis set and according to the
variation principle, these are the best energies for the given basis set. The corresponding values
of the coefficients are then obtained by solving the secular equations using the two energies: the
lower energy gives the coefficients for the bonding MO, the upper energy those for the
antibonding MO. The secular equations only give expressions for the ratio of the coefficients in
each case, their individual values are obtained by normalizing the wavefunction.
1 2
2 2 2
= + + = +
}
S c c c c d
B A B A
t (2.1.14)
The complete solutions are a bit cumbersome:
( ) ( )
( )
( ) ( )
( )
( ) ( ) ( )( ) { }
2
1
2 2
2
1
2 2 2
2
2
/
4
1
2
1
1
ES E S E ES C
C ES c
S S B
S A
S B A E
A A
A
B A B A B A
B A
+ =
=
)
`
+ + + =
+ =
=
| o o |
|
| o o | o o o o
| o o
(2.1.15)
For two cases the solutions are simple. First when the two atoms are the same, and we can write
o o o = =
B A
. Then the solutions are:
( ) ( ) ( ) { }
( ) ( ) ( ) { }
A B A g antibondin
A B A bonding
c c S c S E
c c S c S E
= = =
= + = + + =
, 1 2 1 , 1
, 1 2 1 , 1
2
1
2
1
| o
| o
(2.1.16)
- 27 -
In this case the best bonding function has the form
B A
+ + + and the corresponding antibonding
function is
B A
+ + , in agreement with the earlier intuitive discussion on the hydrogen
molecule-ion. However, we can now explore how the energies and composition of the MOs
change with the parameters | and S. The easiest way of doing so is to suppose that | is
proportional to S, so S k | = , k being a constant. This is illustrated in Fig. 2.1.14.
The other extreme case, when the energies of the AOs are widely different, supposes that
| o o >>
B A
. Then the solutions (with
A
+ taken as the MO with the lowest energy) are
{ } ( ) ( )( ) { }
( )( )
{ } ( ) ( )( )
( )( ) { }
{ } ( )
A B B A A
B A B B
B A B A B g antibondin
B A A B
B A A A A bonding
S S X
S S c
S c S S X E
S c
S S c S X E
o o o o |o |
o o | o |
o o | o | | o
o o | o |
o o | o | o
+ =
+ =
= + =
=
= =
2 2
2
2 2
2
2 2
2
1
; ; 1 2
; 1 ; 1
(2.1.17)
Fig. 2.1.15 shows the effect of increasing
B A
o o on the bonding and antibonding energies,
calculated from eqs. (2.1.17). When the energy separation is very large (as in
(1s,2s)-overlap of homonuclear diatomics), then the bonding orbital has 1 =
A
c corresponding to
pure
A
+ , and the antibonding MO has 1 =
B
c , so that it is pure
B
+ . This justifies the neglect of
(1s
A
,2s
B
)-overlap in homonuclear diatomic molecules, because the two orbitals remain
effectively unchanged in the molecule. A classical analogue is the coupling between two
pendulums sharing a common support. When they have very different frequencies they barely
Fig. 2.1.14. The dependence of the
bonding and antibonding energies on
the overlap, calculated using the
expressions 1.22 and making the
assumption that the resonance
integral is proportional to the
overlap, S k | = .
Fig.2.1.15. The dependence of the
bonding and antibonding orbital
energies on the energy separation of
the contributing atomic orbitals,
calculated from eqs. (2.1.17)
- 28 -
affect each other and swing at their own natural frequencies. In contrast, when the frequencies
are similar they interact strongly and exchange energy freely.
In general it is now clear how the LCAO coefficients for heteronuclear diatomics are
found. One chooses a basis set, solves the secular equations for the energies and then finds the
optimal coefficients. There is still the problem of finding the Coulomb and resonance integrals.
One approach is to estimate them from spectroscopic experiments. This combination of
empirical data and quantum mechanical calculations has given rise to the semi-empirical
methods of molecular structure calculation. The modern tendency , however, particularly for
small molecules, but increasingly for bigger ones too, is to calculate energies and wavefunctions
from first principles. These give rise to ab initio methods, which are now widely used in
quantum chemistry.
2.1.7 Hybridization and The Structure of Polyatomic Molecules.
(a) Hybridization
When dealing with heteronuclear diatomics there is often no clear-cut distinction between
the energies of the diatomic orbitals, and therefore no precise criterion about which AOs should
be combined to an MO. This is illustrated for LiH in Fig. 2.1.16
Although Li2s lies closer to H1s in energy, the Li2p orbitals are not far away and cannot be
ignored. While the minimal basis set is (HJ1s, Li2s), a better description will be obtained if it is
enlarged to (H1s, Li2s, Li2p) and the MOs are expressed as linear combinations of all three. A
variational calculation then gives: s H p Li s Li 1 87 . 0 2 29 . 0 2 41 . 0 + + = + as the optimum
wavefunction with this basisset, demonstrating that the 2p-AOs make a substantial contribution.
At this stage we seem to have lost an attractive feature of the LCAO-MO theory, for now it
seems to be impossible to view the Li-H bond as being formed from the overlap of two AOs,
one on Li and the other on H. A way out is to regard the MO as being arising from the overlap of
H1s and a hybrid orbital on Li, consisting of a linear combination of Li2s and Li2p. The MO can
be expressed then alternatively as s H Lihybride 1 87 . 0 5 . 0 + = + with
p Li s Li Lihybride 2 58 . 0 2 81 . 0 + = . Thus, by constructing hybrids we retain the simplistic picture
of bonding, but with a more complicated atomic contribution. In a sense, hybridization is
Fig. 2.1.16. Hydrogen and lithium
atomic energy levels: H1s overlaps
with both Li2s and Li2p, and the
resulting MO can be viewed as arising
from the overlap of H1s with a
(Li2s,Li2p)-hybrid orbital. Li1s is a
core orbital and plays only a minor
role in the bonding.
- 29 -
unnecessary, there is no compelling physical or mathematical reason. Nevertheless, hybridization
may help to disentangle the various contributions to the energies of bonds, as we will see by
considering LiH more closely.
The shape of the Li hybrid is shown in Fig. 2.1.17. The bulk of the amplitude lies in the
internuclear region. This distortion arises from the interference between the Li2s and Li2p
AOs, because at one side of the nucleus (where their amplitudes have the same sign) they add,
but they partially cancel at the other side (where their signs are opposite). The distortion gives
rise to a stronger bond because the overlap with H1s is enhanced. Of course, exactly the same
enhancement is present in the three AO-description of the bond, but hybridization focused our
attention on it.
Why, though, does the Li-atom hybridize as far as p Li s Li 2 58 . 0 2 81 . 0 + and not further to
for instance p Li s Li 2 71 . 0 2 71 . 0 + , which is even more strongly distorted and would give an even
better overlap? The reason lies in an opposing contribution to the final energy of the molecule.
The actual hybrid is 66% Li2s and 34%Li2p and so an electron in it can be thought of as
spending 34% of its time in an excited state. In the alternative hybrid this would be 50%.
Therefore, in order to achieve better overlap, it
is necessary to invest more energy in the promotion of the Li-atom to a higher energy
configuration. Obviously the two effects are in competition and the outcome, the observed
hybrid is a compromise. Its precise composition, and the optimum energy of the molecule, may
be obtained by calculation of the integrals that occur in the expressions obtained from the
variation principle.
(b) The structure of polyatomic molecules
Polyatomic molecules are molecules consisting of more than two atoms, and include most of the
interesting molecules for chemistry and biology. Their bonds are built in the same way as in
diatomic molecules: AOs overlap and give rise to MOs spreading over all the atoms. But why
do they adopt their characteristic shapes? Why is H
2
O triangular, NH
3
pyramidal, and CH4
Fig. 2.1.17. (a) A cross-section through the (Li2s, Li2p)-hybrid showing the accumulation of
amplitude on one side of the nucleus. (b) The H1s-orbital overlaps the hybrid strongly, and a
stronger bond is formed than with Li2s alone.
- 30 -
tetrahedral, why are the atoms involved in the peptide bond all positioned in the same plane? The
shape of H
2
O will illustrate the approach. The ground state configuration of the O-atom is
1 1 2 2 2
2 2 2 2 1
y x z
p p p s s . This suggests a minimal basis set of ( )
B A y x
s H s H p O p O 1 , 1 , 2 , 2 , with four
electrons to accommodate in the bonds (two from O, one from each H). MOs can be formed by
overlapping each H1s with an O2p, forming two o-bonds, Fig. 2.1.18. Each bonding MO
accommodates two electrons, and so all four are accounted for by the two o-bonds. The
configuration of H
2
O is therefore
2 2 2 2 2
2 2 1
B A z
p s s o o . Since the O2p-AOs are perpendicular, this
predicts a 90
o
H-O-H bond angle. This simple picture is quite good, but an important aspect is
missing: the H
2
O molecule is bent, the real bond angle is 104.45
o
, and this discrepancy must be
explained. The bond angle will increase when the basis set is increased to include the O2s-
orbital, which is nearby in energy and cannot be ignored. The molecular energy can be calculated
using this extended basis set and the variation principle, and the value can plotted as a
function of the bond angle. The minimum energy of the molecule turns out to occur when the
bond angle is about 104
o
, in agreement with observation. However, this does not give much
insight, and the best way of seeing how the inclusion of O2s opens the bond angle is to use the
language of hybridization.
Fig. 2.1.18. A primitive description of the
electronic structure of water. The H1s AOs
overlap the O2p AOs and result in a 90
o
molecule. When O2s is included, the bond
angle increases to its observed value of 104
o
.
- 31 -
(c) Orthogonality and Hybridization
In the MO description of H
2
O we aim to construct two O-H bonds that are chemically
equivalent, but spatially distinct.
How does this apply to H
2
O? Ignoring the O2s contribution, there is no promotion energy, and
the two bonds (involving only
x
p and
y
p ) have a moderately good (s,p)-overlap. When O2s-
hybridization is allowed, forming the two equivalent hybrids h and h, the bond strength
increases because the overlap improves, but a promotion energy is required because the 2s-
electrons now take part in the bonding. The actual shape of the molecule, which is found by
minimizing the total energy is a compromise between strong bonding and promotion energy and
Fig. 2.1.19 (a) p and p can be
expressed as linear combinations of
p
x
and p
y
but the combinations are
not orthogonal. (b) the orthogonal
hybrids, h and h, obtained by
mixing 2s-character into p and p.
Fig. 2.1.20 Three orthogonal AOs
hybridize to give three orthogonal
hybrids. While the first two are
chemically equivalent and each bind an
H-atom, the third (dark) is different and
in the case of H
2
O contains 60% 2s-
character.
- 32 -
corresponds to a bond angle between these extremes (in reality 104.45
o
). Finally, the electronic
configuration of the H
2
O molecule is given by
2 2 2
1 , '
2
1 ,
2
2 ' ' 1
O B A
z s h s h o
p h s o o . The two electron pairs
that are put in the third hybrid ' ' h and
O
z
p 2 are called lone pairs.
Various forms of hybridization play a decisive role in determining the shapes of
molecules. In a number of important molecules (for instance NH
3
and CH
4
), there are more than
two equivalent bonds. For instance three equivalent hybrids may be formed from the basis (2s,
2p
x
, 2p
y
). This is just a special case of the H
2
O calculation with u=120
o
, and we find three
hybrids of the general form ( ) 3 2 2 p s + directed towards the corners of an equilateral
triangle, Fig. 2.1.21.
( )
( )
( )
( )
y x
iii sp
y x
ii sp
x
i sp
p p s
p p s
p s
2
2
1
2
6
1
2
3
1
2
2
1
2
6
1
2
3
1
2 2 2
3
1
2
2
2
= +
+ = +
+ = +
(2.1.18)
This type of hybridization is called
2
sp -hybridization and occurs for instance for the carbon
atoms in the ring of benzene. Note that these
2
sp -wavefunctions are normalized and orthogonal.
For a molecule like BeH
2
the H-Be-H bond is 180
o
. The ground state electronic
configuration of the Be-atom is
2 2
2 1 s s . To represent the directionality of the BeH
2
bonds and
the fact that they are equivalent, two hybrid orbitals of the be atom are formed by taking a linear
combination of the 2s orbital and one of the 2p orbitals. Of course energy is required to excite
the 2s electrons into the 2p AO, but more than enough energy to do this is regained when a stable
chemical compound with two bonds is formed. The two so-called sp-hybrids formed this way are
( )
( )
) 2 2 (
2
1
) 2 2 (
2
1
x ii sp
x i sp
p s
p s
= +
+ = +
(2.1.19)
- 33 -
Carbon atoms have the electronic configuration
2 2 2
2 2 1 p s s , and the outer four valence
electrons may be used to form sp
3
hybrid orbitals:
( )
( )
( )
( )
( )
( )
( )
( )
z y x
i sp
z y x
iii sp
z y x
ii sp
z y x
i sp
p p p s
p p p s
p p p s
p p p s
2 2 2 2
4
1
2 2 2 2
4
1
2 2 2 2
4
1
2 2 2 2
4
1
3
3
3
3
+ = +
+ = +
+ = +
+ + + = +
(2.1.20)
These orthonormal orbitals point in the directions shown in Fig. 2.1.22 in agreement with the
tetrahedral structure of CH
4
and with the geometries of the alkanes. The four electrons in 2s and
2p are put in the four hybrid sp
3
orbitals and each can form a o bond with hydrogen or another
element. For other elements further hybridization schemes involving d-orbitals can be used to
Fig.2.1.21. Three important types of
symmetrical equivalent hybrid orbitals: (a)
linear (180
o
) sp-hybrids, (b) plane
triangular (120
o
) sp
2
-hybrids and (c)
tetrahedral (109
o
) sp
3
-hybrids.
- 34 -
account for bipyramidal structures (coordination nr 5) and octahedral structures (coordination nr
6).
When we come to NH
3
we must apply the idea of the lone pair electrons, already introduced for
H
2
O. We use the
1 1 1 2
2 2 2 2
z y x
p p p s configuration on N to construct four sp
3
hybrids just as in
carbon. Now we must put two valence electrons of N in one of these hybrids, leaving three
hybrids available to form bonds with the hydrogens. This leads to a tetrahedral structure of NH
3
(bond angle 109
o
) with one apex of the tetrahedron containing a lone electron pair, as shown in
fig.2.1.23. (The experimentally observed bond angle is 107
o
). These lone pair electrons are
available for bonding to, for example a proton, H
+
, to form NH
4
+
.
Fig. 2.1.22. Directions of the sp
3
hybrids formed from the 2s and
three 2p orbitals.
- 35 -
Although we have discussed the H
2
O molecule as being sp
2
-hybridized, in reality it is often
discussed in much the same manner as NH
3
, except that we now have two lone-pairs as shown in
Fig. 2.1.23. The difference between the predicted and observed bond angle, 109
o
vs. 104
o
, can be
accounted for by adding more terms to the wavefunction. The lone-pairs of H
2
O are available to
form bonds with other atoms. In particular, the interaction between hydrogen atoms on other
water molecules and the lone-pairs gives rise to the hydrogen bond and the properties of water.
3.1.8 Hckel Molecular Orbital Theory
Molecules with extensive t-bonding systems, like benzene or for instance the photosynthetic
pigments chlorophyll a, |-carotene, are not described very well by valence bond theory, because
the t-electrons are often not localized in a single bond, but instead are delocalized over the
whole molecule. Their electronic structure can be described in a simple but approximate
molecular orbital method developed by Hckel in 1930. The two types of bonds involved in
these molecules are illustrated in Fig. 2.1.24 for ethylene (C
2
H
4
).
Fig. 2.1.23. (a) The configuration of
NH
3
, including the orbital with the
lone-pair. (b) H
2
O plus its two lone-
pairs.
- 36 -
Ethylene is a planar molecule and the bonds between the carbon and hydrogen atoms in the plane
are formed from the sp
2
-orbitals of the two carbons and the 1s-orbitals of the four hydrogens.
This forms the o-bond framework shown for ethylene in Fig. 2.1.24a. The 2p
z
orbitals of the
carbon atoms, that are not involved in the o-framework, overlap sideways, forming the t-system.
The contribution to the bonding energy due to the two electrons in the t-orbital is sufficient to
prevent the molecule from distorting into a non-planar configuration. So this simple picture
explains the bond angles in ethylene and its planar structure.
Hckel molecular orbital theory assumes that the t electrons, which are responsible for
the special properties of conjugated and aromatic hydrocarbons, do not interact with one another,
and so the many-electron wavefunction is just a product of one-electron molecular orbitals.
Furthermore it assumes that the structure of the molecule is given by the o-framework. The
molecular orbital of the delocalized t orbital of ethylene is then represented by:
( ) ( )
2 2 1 1
2 2
z z
p c p c + = +
t
(2.1.21)
where
2 1
2 , 2
z z
p p are the 2p orbitals on the two carbon atoms not used in forming the sp
2
hybrids. The corresponding secular determinant is:
0
22 21 21
12 12 11
=
E ES
ES E
o |
| o
(2.1.22)
where of course
zj zi ij zj zi ij zi zi ii
p p S j i p H p p H p 2 2 , , 2 2 , 2 2 = = = = | o . In Hckel
molecular orbital theory, the secular determinant is simplified by making the following
assumptions:
1. All overlap integrals
ij
S are set to zero. This may be reasonable for
z
p 2 orbitals on
neighboring atoms, but certainly a rather crude approximation. It can easily be corrected!
Fig.2.1.24 Bonding in ethylene. (a) In the
plane of the nuclei: the formation of a o
bond between the carbon atoms 1 and 2
using sp
2
hybrid orbitals and o-bonds
between the 4 H 1s electrons and the
remaining sp
2
orbitals on each C-atom. (b)
Perpendicular to the plane of the 6 nuclei:
formation of a t-bond between the two 2p-
orbitals (that were not involved in the sp
2
-
hybrids.
- 37 -
2. All diagonal terms are assumed to be identical: i
ii
= , o o . For ethylene this is perfect.
For longer conjugated molecules, the atoms in the middle are different from those at the
ends. Also more complex conjugated molecules may contain different atoms (like N, O).
3. The resonance integrals
ij
| are all set equal to | for neighboring atoms and zero for non-
neighbors. This is justified by the fact that the spatial overlap for between two 2p
z
orbitals
on non-neighbors will be very small.
With these assumptions the secular determinant for the t-electrons in ethylene becomes
0 =
E
E
o |
| o
(2.1.23)
In Hckel theory the Coulomb integral o and the resonance integral | are regarded as empirical
parameters to be evaluated from experimental data on the molecule. Thus in Hckel theory it is
unnecessary to specify the Hamiltonian operator (although it is instructive to do so!).
Equation 2.1.23 can easily be solved for the energy to obtain | o = E (like the solution
for the H
2
+
molecule (but note that the quantities o, | now are completely different). Thus there
are bonding and antibonding orbitals, as shown in Fig.2.1.25. The resonance integral is negative
and so the energy of the lowest level is | o + . The bonding t-orbital is occupied by an electron
pair composed of the two remaining electrons of the carbon atoms, and so the t-electronic
energy of ethylene is | o 2 2 + . The wavefunctions for the bonding and antibonding t-orbitals
are obtained by substituting the obtained bonding and antibonding energies into the original
secular equations:
( )
( ) 0
0
2 1
2 1
= +
= +
E c c
c E c
o |
| o
(2.1.24)
Substituting | o + = E in either equation yields
2 1
c c = so that the wavefunction for the bonding
t-orbital is
Fig.2.1.25. Hckel molecular
orbitals for ethylene. The
carbon nuclei are represented
by dots, and the nodal planes
for the MOs are represented
by the dashed lines.
- 38 -
( )
2 1 1
2 2
2
1
z z
p p + = +
t
(2.1.25)
The bonding t-orbital is shown in Fig 2.1.25 alongside the energy level.
( )
2 1 1
2 2
2
1
z z
p p = +
t
(2.1.26)
The antibonding t-orbital is also shown in Fig.2.1.25. The Hckel molecular orbital theory gives
us an estimate of the excitation energy to the first excited state of ethylene. It is evident from
Fig.2.1.25 that this excitation energy is | 2 . This figure also gives us the opportunity to
introduce some useful nomenclature. The highest occupied molecular orbital is called HOMO,
the lowest unoccupied molecular orbital is called LUMO.
The t-electronic energy of the planar molecule 1,3-butadiene (CH2=CHCH=CH2) is
readily calculated using Hckel theory. The secular determinant is:
0
0 0
0
0
0 0
=
E
E
E
E
o |
| o |
| o |
| o
(2.1.27)
(Note that now the two outer carbon atoms are in fact different form the inner two!). If | is
factored from each column and ( ) | o / E is replaced by x we obtain
0
1 0 0
1 1 0
0 1 1
0 0 1
=
x
x
x
x
or 0 1 3
2 4
= + x x (2.1.28)
so that 618 . 1 , 618 . 0 = x . Thus, for 1,3-butadiene there are four possible energy levels for the
t-electrons, two bonding and two antibonding as shown in Fig.2.1.26.
| o
| o
| o
| o
t
t
t
t
618 . 1
618 . 0
618 . 0
618 . 1
* 4
* 3
2
1
=
=
+ =
+ =
E
E
E
E
(2.1.29)
For each carbon atom of 1,3-butadiene two electrons are in an 1s-atomic orbital, three electrons
are put in the three sp
2
-hybrids to form o-bonds with neighboring C, H atoms. Consequently on
each carbon atom there is one electron left, four in total, to fill the t-orbitals. The 2t-orbital is
the HOMO, the 3t* the LUMO of butadiene.
By substitution of the four energies into the secular equations the four Hckel molecular orbitals
of 1,3 butadiene are found
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )
4 3 2 1 * 4
4 3 2 1 * 3
4 3 2 1 2
4 3 2 1 1
2 372 . 0 2 602 . 0 2 602 . 0 2 372 . 0
2 602 . 0 2 372 . 0 2 372 . 0 2 602 . 0
2 602 . 0 2 372 . 0 2 372 . 0 2 602 . 0
2 372 . 0 2 602 . 0 2 602 . 0 2 372 . 0
z z z z
z z z z
z z z z
z z z z
p p p p
p p p p
p p p p
p p p p
+ = +
+ = +
+ = +
+ + + = +
t
t
t
t
(2.1.30)
- 39 -
These four t-orbitals are schematically drawn in Fig.2.1.26. Notice that the t-orbitals extend the
entire length of the molecule. Note also the increasing number of nodal planes perpendicular to
the xy-plane, reflecting the decrease in bonding between
t 1
+ and
* 4t
+ . How does the
delocalization of the t-electrons over the whole molecule affect the total bonding energy of the
molecule? Naively one might have thought of 1,3-butadiene as two covalently-linked ethylene
molecules with one pair of t-electrons between the first two carbon atoms and the second pair of
t-electrons between the last two carbon atoms. The total bonding energy of the t-electrons
would then have been 4o+4|. In reality the total bonding energy is 2(o+1.618|) + 2(o+0.618|)=
4o+4.472|. Thus an amount of 0.472| is gained by delocalizing the electrons over the whole
molecule. Finally the excitation energy is | 236 . 1 , which corresponds to about 300 nm. When
compared to ethylene, 1,3-butadiene absorbs clearly at lower energy than ethylene. This is
typical for linear-chain conjugated molecules. Carotenoids are natural occurring pigments that
play a role in photosynthesis and photoprotection. They contain between 18 and 26 conjugated
carbon atoms and typically absorb light between 500 and 600 nm.
The benzene molecule (C
6
H
6
) is ring-shaped with 120
o
angles between each C-triplet in
the ring. Each C-atom is sp
2
-hybridized and o-bonds are formed with sp
2
-hybrids of the
Fig.2.1.26.
Hckel molecular orbitals
for 1.3-butadiene.
The orbitals are viewed
perpendicular to the plane
of the molecule.
The carbon nuclei are
represented by dots, and
the nodal planes for the
MOs are represented by
the dashed lines.
With each carbon atom
contributing one electron
the 1t and 2t orbitals are
filled.
- 40 -
neighboring C-atoms and the 1s of the H-atom. The remaining six 2p
z
electrons will be used to
fill t-orbitals that spread out over the whole benzene molecule. A trial wave function for the t-
electrons is:
zi
i
i
p c 2
6 1
=
= +
t
Then the Hckel secular determinant for benzene becomes
0
0 0 0
0 0 0
0 0 0
0 0 0
0 0 0
0 0 0
=
E
E
E
E
E
E
o | |
| o |
| o |
| o |
| o |
| | o
(2.1.31)
The six roots are:
| o
| o
| o
| o
t
t t
t t
t
2
2
* 6
* 5 * 4
3 2
1
=
= =
+ = =
+ =
E
E E
E E
E
(2.1.32)
Note that two of the t-energies are degenerate, which relates to the circular symmetry of the
molecule. Using benzenes six 2p-electrons, pairs of electrons go into the lowest three energy
orbitals. Thus the t-electronic energy of benzene is
( ) . 8 6 4 ) 2 ( 2 | o | o | o
t
+ = + + + = E (2.1.33)
The equations for the six Hckel molecular orbitals for benzene are not given here, but the
corresponding electron densities are shown in Fig. 2.1.27.
Note that the total p-electronic energy is more negative than three times the value of
ethylene ( ) | o 6 6 + indicating that C
6
H
6
is not just a molecule with three double bonds. Again
the additional contribution to the bonding energy arises from allowing the t-electrons to move
freely over the whole molecule.
The Hckel theory is an example of a semi-empirical molecular orbital method. We have
used a simple Hamiltonian (neglecting many terms) to find the molecular orbitals and their
energies. We now can use experimental quantities to fit o and | for ethylene. We then use these
values to make predictions for butadiene, benzene and so on. This method does not give
quantitative results, but it does provide us with qualitative insights about larger systems for
which the more computationally intensive methods are too costly or time-consuming, and it does
give us insight into the electronic excited states of conjugated t-electron molecules.
- 41 -
2.1.9 Symmetry
Formaldehyde, see transition moments
Fig.2.1.27. Hckel molecular orbitals for benzene. The carbon atoms are represented by dots,
the nodal planes perpendicular to the molecular plane are represented by dashed lines.
Exercise: Calculate the Hckel molecular orbital energies and molecular orbitals of the planar
radical ( i.e. 1. nonpaired electron) CH
2
CHCH
2
.
Exercise: Calculate the Hckel molecular orbital energies and molecular orbitals for the
molecule cyclo-butadiene (C
4
H
4
)
- 42 -
2.1.10 Quantum Chemistry
Configuration Interaction, Hartree-Fock, DFT
2.2 Non-Covalent Interactions.
So far we have discussed the covalent interactions that occur in the bond formation of molecules.
However, in proteins, membranes and nucleic acids interactions between remote parts of the
molecule and between the molecule and its surroundings determine their three dimensional
structure. When two neutral molecules come close to one another (even when they are non-polar)
the various interactions between the electrons and nuclei of one molecule and the electrons and
nuclei of the other produce a potential energy of interaction. At very short distances the
molecules repel each other strongly. At certain intermediate distances, the potential energy is
negative, the molecules attract each other, but usually weakly, of the order of kT . At very large
distances the potential energy approaches zero. Thus if we plot the interaction energy as a
function of distance we obtain curves like Fig.2.2.1. Here we will discuss three of such
interactions: the interaction between dipoles etc., the Van der Waals forces and the hydrogen
bond.
.
2.2.1 Charge-Charge and dipole-dipole interactions.
When two molecules which carry charges come close together their interaction energy is
given by Coulombs law. The potential energy falls off as 1/r and hence it is referred to as a long
range interaction. In water, which is the appropriate medium inside cells, the Coulomb
interactions are reduced by a factor, which is the dielectric constant of water. This dielectric
constant is large, 80 =
water
c and originates from the fact that water molecules have a large
Fig.2.2.1 The energy of interaction of two N
2
-molecules as a function of distance
according to the Lennard-Jones potential. Note that the ordinate is in Kelvins; the
interaction energy has been divided by the Boltzmann constant.
- 43 -
permanent dipole which effectively screens the charges. In a protein environment 2 ~ c and ion
pairs with opposite charges may contribute significantly to the stabilization of a biomolecular
structure. Of course on an atomic scale the concept of the dielectric constant becomes
meaningless and one would have to calculate explicitly all the interactions between the charges.
However, even in the absence of net charges, molecules attract each other. One example
concerns polar molecules with a very asymmetric charge distribution. When two of such
molecules are close together, their dipoles
2 1
, interact and this interaction
a b
affects their potential energy. If the molecules would take up all relative orientations with equal
probability then the average interaction energy would be zero because the attractive interaction
(occurring when the dipoles are head-to-tail) is cancelled by the repulsive interaction (when the
dipoles are head-to-head) (see Fig. 2.2.2). However, in a fluid the attractive orientations are more
likely to occur than the repulsive, because they correspond to a lower energy.
To get the correct form of the final expression we note that the interaction energy of two polar
molecules rotating at some fixed separation r is given by:
( ) f
r r
V
3
0
2 1
2 1
3
0
2 1
4
cos cos 3 cos
4 tc
| | u
tc
= = , (2.2.1)
where f is called the orientation factor.
The average interaction energy is given by: f
r
V
3
0
2 1
4tc
= (2.2.2)
where f now includes a weighting factor in the averaging that is equal to the probability that
the molecules (dipoles) will adapt a certain orientation. This probability is given by the
Boltzmann factor
kT E
e p , with E interpreted as the potential energy of interaction of the two
dipoles in that orientation. This implies that for every orientation f we have:
kT f V
e f p
) (
) (
= with f
r
f V
3
0
2 1
4
) (
tc
= . (2.2.3)
Fig 2.2.2 (a) Attractive
dipole-dipole interaction
between two molecules.
(b) repulsive counterpart
of (a).
- 44 -
When the potential energy of interaction between the two dipoles is very small compared with
the energy of thermal motion, we can use kT V << , expand the exponential function in p and
retain only the first two terms:
kT V p 1 (2.2.4)
The weighted average of f is therefore:
.....
4
0
2
3
0
2 1
0
+ = f
kTr
f f
tc
, (2.2.5)
where .. refers to an unweighted spherical average. The average value of f is zero so the first
term vanishes. However, the average value of
2
f is nonzero, because
2
f is positive for all
possible orientations. Thus we can write:
( )
0
2
6 2
0
2
2
2
1
4
f
kTr
V
tc
= (2.2.6)
Since the average value of
2
f turns out to be 2/3, the result is:
2
0
2 1
6
4 3
2
|
|
.
|
\
|
=
tc
kTR
V (2.2.7)
The important feature is the dependence of this energy on the inverse sixth power of the
separation R, and its inverse dependence on the temperature. The latter reflects the fact that the
greater thermal motion overcomes the mutual orientating effects of the dipoles at higher
temperatures.
We will not further discuss this type of interaction. Let it suffice to say that a typical
electrostatic calculation of a protein puts partial charges on all of the atoms (or groups of atoms)
and simply solves the Poisson equation numerically. Note that in a protein, which is a quasi-rigid
structure, the averaging as done above is not valid; this is only allowed for molecules in a freely
moving solvent
2.2.2 Van der Waals forces
The force responsible for the attraction between two non-charged or non-polar
molecules is called the Van der Waals force. The first contribution to the Van der Waals force is
the interaction between permanent dipoles as described above. For freely moving molecules
this contribution is largely cancelled. A second term in the Van der Waals forces arises from the
dipole-induced dipole interaction between molecules. If molecule A has a dipole moment
A
this
creates an electric field which polarizes the charge distribution on molecule B, creating an
induced dipole moment of magnitude
A B
o , where
B
o is called the polarizability of molecule
B. Note that the induced dipole on B is always oriented in the direction of the electric field
- 45 -
created by
A
and consequently the interaction is always attractive. A calculation of this
interaction yields:
( )
6 2
0
2 2
,
4 R
V
B A A B
induceddip dip
tc
o o +
= (2.2.8)
Notice that it also varies as
6
R , simply because the size of the induced dipole on B is
proportional to
A
.
There is a third term varying also as
6
R , which in fact dominates the Van der Waals
forces and was (only) explained in 1930 by London and is therefore called the London force or
London dispersion force. It occurs even when the molecules have no permanent dipole moment.
The abundant evidence for the existence of strong interactions between non-polar molecules is
the formation of condensed phases of non-polar substances such as the condensation of hydrogen
or argon to a liquid at low temperatures and the fact that benzene is a liquid at normal
temperatures. Also in proteins Van der Waals forces and specifically the dispersion force, plays a
major role in the formation of their stable and active conformation, in particular in stabilizing the
hydrophobic core.
The dispersion force originates from the coupling of instantaneous fluctuations in the charge
distribution on one molecule that gives it a dipole briefly; that dipole may induce another dipole
in a neighboring molecule, and these two dipoles will interact favorably. The polarizability of a
molecule is a measure for both of the charge fluctuation in a molecule and for the response to an
applied field; therefore both the initial transient dipole and the response of the neighboring
molecule can be expected to be related to their polarizabilities, and hence that the energy of
interaction should be proportional to
B A
o o . The precise relation we will establish here.
We use perturbation theory to calculate the lowering of the energy when two spherical molecules
are brought to a separation R. The perturbation Hamiltonian is the interaction between two
dipole moment operators centered on the two molecules.
(2.2.9)
, where r is a unit vector along the vector R
connecting the two dipoles. Note that this is of
course the same expression as the one used earlier; now we maintain the dipole moment
operators as vectorial operators.
The unperturbed Hamiltonian is the sum of the electronic Hamiltonians of the two molecules
( )
B A
H H H + =
0
(2.2.10)
The wavefunction of the pair is
B A
n n , where the numbers n
A
, n
B
label the electronic states of
A, B respectively, with for instance
B A
0 , 0 the ground state wavefunction of A, B, resp.
B A
n n is a solution of the unperturbed Schrdinger eq:
( )
( ){ }
B A B A
r r R H tc
- - - = 3 4 1
3
0
1
- 46 -
( ) ( )
B A n n B A B B A A B A B A
n n E E n n H n n H n n H H
B A
+ = + = +
(2.2.11)
We write these joint energies as
B A
n n
E . The ground state is 00 , its energy being
00
E .
The first order interaction is the expectation value of
( ) 1
H
( ) ( )
( )
( ){ }
B A B A
B A B A
R R R
R R R H E
0 0 0 0
3
0
3
0
1 1
3 4 1
0
3 0 4 1 0 0
tc
tc
- - - =
- - - = =
(2.2.12)
where
A A A A
0 0
0
= is the permanent dipole of A and likewise for B. However, dispersion
forces operate for molecules with no permanent dipole; therefore in that case the first order
correction to energy is zero (note that this term survives for polar species). Next we investigate
the second-order correction to the energy. Physically this means that we allow for a distortion of
the molecular wavefunction due to the presence of the other molecule. In perturbation theory this
second order correction is given by:
( )
( )
=
B A
B A
n n
n n
B A B A
E E
H n n n n H
E
,
00
1 1
2
0 0
with 0 , 0 , =
B A
n n (2.2.13)
We write
0 0 00
B A B A
n n n n
E E A + A = , the sum of the excitation energies of the two molecules. The
perturbation Hamiltonian has quite a complicated form. It is easier to handle if expressed in
terms of a system of coordinates in which the interatomic separation defines the z-axis. Then:
( )
( ){ }
Bz Az By Ay Bx Ax
R H tc 2 4 1
3
0
1
+ =
(2.2.14)
Even so, the second-order correction to the energy takes the formidable form:
( )
( )
( )
0 0
,
2
3
0
2
0 2
2 0 4 1
B A
B A
n n Bz Az By Ay Bx Ax B A
B A Bz Az By Ay Bx Ax
n n
n n
n n R E
A + A +
+ =
tc
(2.2.15)
However, most of the nine terms vanish. Consider a term such as
0 0
By Ay B A B A Bx Ax
n n n n .
This includes the factor
A Ay A A Ax A
n n 0 0 , which must be zero in a spherical molecule.
The quickest way to see this is to choose an alternative coordinate system with the x-axis the
same, but the y-axis pointing in the opposite direction. The product then changes sign. But its
contribution to the energy can not depend on our choice of an axis system, therefore it must be
- 47 -
zero. The same argument applies to all the cross terms in the expression for
( ) 2
E , and so the only
terms that survive are the three terms of the form 0 0
Bx Ax B A B A Bx Ax
n n n n etc. In
the case of spherical molecules (and atoms) all three surviving terms are equal and so:
A A A A A A A A A A Ax A
n n n n 0 0
3
1
0 0
- = , and likewise for the other two
components, and for the three components labeled B. As a consequence the entire expression
reduces to a single term:
( )
( )
( )( )
A + A
- -
=
B A
B A
B B A A
n n
n n
n B n B n A n A
R E
,
0 0
0 , 0 , 0 , 0 ,
2
3
0
2
4 1
3
2
tc
(2.2.16)
(with ) 0 , 0 , =
B A
n n
This result confirms that there is a non-zero interaction energy which is attractive (
( ) 2
E
is negative) and depends on the separation of the two molecules as
6
1 R .
We can obtain an approximate, revealing and useful form of this expression by making use of the
so-called closure relation.
( )
( ) ( )( ) { }
( )
2 2
6
2
0
2
,
0 , 0 , 0 , 0 ,
2
3
0
2
1 1
24 1
1
4 1
3
2
B A
B A
n n
n B n B n A n A
B A
R
R E
B A
B B A A
c t
tc
|
|
.
|
\
|
A + A
|
.
|
\
|
~
- -
)
`
A + A
=
(2.2.17)
(with ) 0 , 0 , =
B A
n n . We have used
A n
A
A ~ A
0
and likewise for B,
B A
A A , reflecting the average
energy difference between ground and excited states. Furthermore, 0 0
2 2
A A
= and the same
for B. This relation can be taken further by using the relation between the mean square dipole
moment and the polarizability of a molecule: 2 3
2
A A A
A ~ o , idem for B. Inserting these
expressions yields:
( )
6 2
0
1
4
' '
2
3
R E E
E E
V
B A
B A
B A
disp
tc
o o
|
|
.
|
\
|
+
= (2.2.18)
where
B A
A A , have been put equal to
B A
E E , , the energies of the first electronic transitions of
these molecules, and
B A
' , ' o o are their polarizability volumes (
0
4 ' tc o o = ) .
These three terms add to give the total attractive energy between molecules A and B. In
most cases this energy is on the order of
3
10 2
~ eV at a distance of 0.5 nm. Of course the
larger the dipole moments or the polarizabilities, the stronger is the interaction.
- 48 -
2.2.3. The intermolecular potential energy
At small distances the molecules repel strongly, (we try to push the electrons of one molecule
into the non-bonding orbitals of the other, the bonding orbitals are occupied by electron pairs), so
when this force is added to the attractive forces discussed above we obtain the total
intermolecular potential energy. To represent this as a simple function of R, we take the
repulsion to be proportional to
n
R
where n is fit empirically. One very useful empirical function
is named after J.E. Lennard-Jones and is given by
(
(
|
.
|
\
|
|
.
|
\
|
=
6 12
4
R R
V
o o
c (2.2.19)
In this potential, the 12
th
power dependence for the repulsive energy is chosen for convenience
and because it gives a reasonable fit to the data. The minimum in this potential is at ( ) o
6 1
2 = R
where c = V . Note that 0 = V at o = R , so the energy rises very steeply for small R (see Fig.
2.2.3, where the Lennard-Jones potential was given for two N
2
-molecules). The values of c and
o can be found by fitting experimental data on the second virial coefficient, gas viscosity and
molecular beam scattering cross sections. Typical values for the parameters c and o for
interactions between like atoms and molecules are given in table 2.2.1.
Fig. 2.2.3 Typical form of the Lennard Jones potential. The distance at which the
minimum occurs would be two times the van der Waals radius of the molecules
involved.
The energy of interaction of two N
2
-molecules as a function of
distance according to the Lennard-J ones potential.
(
(
|
.
|
\
|
|
.
|
\
|
=
6 12
4
R R
V
o o
c
( ) o
6 1
2 = R
The Lennard-Jones Potential (1)
- 49 -
K
k
/
c
pm / o
Ar 120 341
Xe 221 410
H
2
37 293
N
2
95.1 370
O
2
118 358
Cl
2
256 440
CO
2
197 430
CH
4
148 382
C
6
H
6
243 860
The pairwise intermolecular potential energy plays a crucial role in molecular dynamics
simulation that attempt to calculate the dynamics of proteins or nucleic acids. Examples are the
binding of an inhibitor by an enzyme and the folding of a peptide chain to form a globular
protein. In such calculations much more complicated equations for the intermolecular potential
energies are used because the 12
th
power dependence in the Lennard-Jones potential is arbitrary.
Pair-potentials based on atom-atom potentials can also be used to calculate molecule-molecule
interactions.
2.2.4. The hydrogen bond
The hydrogen bond plays a crucial role in the structure of biomolecules. Also many of the
remarkable properties of water relate to the formation of hydrogen bonding networks in water.
Fig. 2.2.4 shows again the structure of water, where the Van der Waals radii have now been
added to the atoms. The large electronegativity difference between H and O confers a 33% ionic
character on the OH-bond as reflected by waters dipole of 1.85 Debye units. Water is clearly a
highly polar molecule, a phenomenon with enormous implications for living matter. The
electrostatic interactions between the dipoles of two water molecules tend to orient them such
that the O-H bond on one molecule points towards a lone pair electron cloud on the oxygen atom
of the other water molecule as illustrated in Fig.2.2.5. This results in a directional intermolecular
association known as the hydrogen bond. In general a hydrogen bond may be represented as
A H D , where D-H is a weakly acidic donor group such as N-H or O-H, and A is a
lone-pair bearing atom and thus a weakly basic acceptor atom The peculiar requirement of a H
atom in the A H D interaction stems from the hydrogen atoms small size: only a
hydrogen nucleus can approach the lone-pair electron cloud of an acceptor atom closely enough
to permit an electrostatic interaction of significant magnitude.
Hydrogen bonds are structurally characterized by an A H distance that is at least 0.5
shorter than the calculated van der Waals distance (distance of closest approach between two
non-bonded atoms). In water for example the H O distance is about 1.8 , vs. 2.6 for the
corresponding van der Waals distance. The energy of the hydrogen bond, about 20kJmol
-1
in
H
2
O, is small compared to covalent bond energies (for instance 460 kJmol
-1
for an O-H covalent
bond). Nevertheless, most biological molecules have so many hydrogen bonding groups that
Table 2.2.1. Lennard Jones
potential parameters for
various atoms and molecules.
Note that the depth of the
potential has been expressed as
a temperature by dividing c by
the Boltzmann constant k.
- 50 -
hydrogen bonding is of paramount importance in determining their three-dimensional structures
and their intermolecular associations.
Fig 2.2.4 The structure of the water molecule. Top: The electronic structure of the water
molecule shown as contour maps of electron density through the center of the molecule, viewed
from two perpendicular directions. Bottom: The outline represents the van der Waals envelope
of the molecule, where the attractive components of the van der Waals interactions balance the
repulsive components). The skeletal model of the molecule indicates its covalent bonds
- 51 -
The structure of ice provides a striking example of the cumulative strength and organizing
potential of many hydrogen bonds. X-ray and neutron diffraction studies have established that
water molecules in ice are arranged in an unusually open structure (see Fig.2.2.6). Each water
molecule is tetrahedrically surrounded by four nearest neighbors to which it is hydrogen bonded.
In two of these hydrogen bonds the central H
2
O molecule is the donor and in the other two it is
the acceptor. As a consequence of its open structure, water is one of the very few substances
that expands upon freezing (at 0
o
C, liquid water has a density of 1.00 g.mL
-1
, whereas ice has a
density of 0.92 g.ml
-1
).
Fig. 2.2.5.A hydrogen bond between two water molecules. The strength of the
interaction is maximal when the O-H covalent bond points directly along a lone-pair
electron cloud of the oxygen atom to which its hydrogen bonded.
- 52 -
2.2.5 The hydrophobic interaction
The interactions between water and non-polar surfaces are particularly unfavorable and water is
a poor solvent for non-polar molecules. Non-polar molecules can not participate in the H-
bonding that appears so important in liquid water and aqueous solutions of such molecules have
anomalous physical properties. If dispersed in water, they force the adjacent water molecules to
reorganize in ice-like cages that surround the hydrophobic molecule (see fig 2.2.7).
Because these cage structures are more ordered than the surrounding water, their formation
increases the free energy. This free energy cost is minimized, however, if the hydrophobic (or
hydrophobic parts of amphipathic molecules) cluster together so that the smallest number of
water molecules is affected.
Fig 2.2.6. The structure of ice.
The tetrahedrical arrangement
of the water molecules is a
consequence of the roughly
tetrahedrical disposition of
each oxygen atoms sp
3
-
hybridized bonding and lone-
pair orbitals. Oxygen and
hydrogen atoms are
represented, resp., by red and
white spheres, and the hydrogen
bonds are indicated by the
dashed lines. Note the open
structure that gives ice its low
density relative to liquid water.
- 53 -
Below (fig 2.2.8) we show the phase diagram for the transfer of the nonpolar molecule
cyclohexane to water. The left part is at 20
o
C, the right part at 140
o
C. Note the difference in
entropy change going from liquid cyclohexane to cyclohexane dissolved in water between 20
o
and 140
o
. This of course is related to the ice-like structures that water must form at 20
o
to
accommodate the cyclohexane molecule, which are no longer formed at 140
o
.
Fig 2.2.7 How hydrophilic and hydrophobic molecules interact differently with water.
Fig. 2.2.8 Phase diagram for the transfer of cyclohexane between the gas, liquid and solid
phases and aqueous solution at two temperatures: 20
o
and 140
o
.
- 54 -
Below in fig 2.2.9 we give two examples where the hydrophobic interaction plays a dominant
role in forming a very special structure: the phosphopholipid bilayer and the folded protein. In
both cases is the exposure of hydrophobic surface to water minimized.
Fig 2.2.9 Top: Self-organization of biological molecules driven by the hydrophobic force. Top
Packing arrangement of lipid molecules in an aqueous environment. Lipids are molecules with a
polar head and a hydrophobic tail (often composed of a saturated hydrocarbon). Depending on
their shape they form curved membranes (top left) or flat membranes (top right). Bottom:
Folding of a small water-soluble protein where the hydrophobic residues are now buried inside
and the hydrophilic residues are exposed to the polar solvent.
- 55 -
Note in figure 2.2.9 how the polar amino acid side chains tend to gather on the outside of the
protein, where they can interact with water; nonpolar amino acid side chains are buried on the
inside to form a tightly packed hydrophobic core of atoms that are hidden from water.
- 56 -
4. Macromolecules and Biomolecules
There are macromolecules everywhere, inside us and outside us. Some are natural, they include
polysaccharides such as cellulose, polypeptides such as protein enzymes, and polynucleotides,
such as deoxyribonucleic acid (DNA). Others are synthetic: they include polymers such as nylon,
polystyrene, conducting polymers, dendritic structures etc. that are manufactured by stringing
together and (in some cases) cross-linking smaller units known as monomers. Many (all)
biomolecules including complete genomes can now be chemically synthesized, we are only one
step away from artificial life (synthetic biology)!!!
Life in all its forms, from is the physics and chemistry of macromolecules. Macromolecules give
rise to special problems that include the shapes and the lengths of polymer chains, the
determination of their sizes, and the large deviations from ideality of their solutions. Natural
macromolecules differ in certain respects from synthetic macromolecules, particularly in their
composition and resulting structure, but at the same time the two share a number of common
properties. We concentrate on these common properties here, Another level of complexity arises
when small molecules aggregate into larger particles in a process that is called self-assembly
and gives rise to aggregates. One example is the assembly of hemoglobin from four myoglobin-
like polypeptides. Hemoglobin has new properties that were not present in the individual
monomers, but emerged upon forming the aggregate. A similar type of aggregation gives rise
to a variety of disperse phases, which include colloids. The properties of the disperse phases
resemble to a certain extent the properties of solutions of macromolecules, and we will describe
their common attributes.
4.1 Structure and Dynamics.
The concept of the structure of a macromolecule takes on different meanings at the different
levels at which we think about the arrangement of the chain or network of monomers. The term
configuration refers to structural features that can be changed only by breaking existing chemical
bonds and forming new ones. Thus the chains A-B-C- and -A-C-B- have different
configurations. The term conformation refers to the spatial arrangement of the different parts of a
chain, and one conformation can be changed into another by rotating part of a chain around a
bond.
4.1.1 The different levels of structure.
The primary structure of a macromolecule is the sequence of small molecular residues that make
up the polymer. The residues may form either a chain, as in polyethylene, or a more complex
network, in which cross-links connect different chains as in cross-linked polyacrylamide. In a
synthetic polymer, virtually all the residues are identical and it is sufficient to name the monomer
used in synthesis. Thus the repeating unit in polyethylene is CH
2
CH
2
- and the primary structure
of the chain is denoted as ( CH
2
CH
2
)
n
-.
The concept of primary structure ceases to be trivial in the case of synthetic copolymers and
biological macromolecules, for in general these substances are chains formed from different
molecules. For example, proteins are polypeptides formed from different amino acids (20 occur
naturally) strung together by the peptide bond, -CONH- (see below). The determination of the
- 57 -
primary structure is a highly complex problem of chemical analysis called sequencing. The
degradation of a polymer is a disruption of its primary structure, when the chain breaks into
shorter components.
The secondary structure of a macromolecule is the (often local) spatial arrangement of a chain.
The secondary structure of an isolated molecule of polyethylene is a random coil, whereas that of
a protein is a highly organized arrangement determined largely by hydrophobicity and hydrogen
bonds, and taking the form of random coils, helices (see fig 4.1) and sheets in various segments
of the molecule. The loss of secondary structure is called denaturation. When the hydrogen
bonds in a protein are destroyed (for instance by heating, as when cooking an egg, or by
detergents) the structure denatures into a random coil.
The tertiary structure is the overall three-dimensional structure of a macromolecule. For instance,
the protein shown in Fig 4.1 has helices, sheets and stretches of random coil. The pieces of the
secondary structure interact to form a compact tertiary structure.
A quarternary structure of a macromolecule is the manner in which large molecules are formed
by the aggregation of others Fig 4.2 shows how four molecular subunits, each with a specific
tertiary structure aggregate together. Quarternary structure can be very important in biology. For
instance, the oxygen transport protein hemoglobin consists of four myoglobin-like subunits that
work together to take up and release O
2
.
Fig 4.1 View from two sides of a
small protein, the SH2 domain, part
of the Src protein, a kinase,
meaning that it is involved in signal
transduction via the
phosphorylation of a partner
protein. The SH2 domain regulates
this process. Left: backbone; Right:
Ribbons representing elements of
secondary structure. The protein is
folded into helices (violet, yellow),
sheets (blue, green) and random
coils, connecting the sheets/helices.
- 58 -
4.2 Random coils
The most likely conformation of a chain of identical subunits not capable of forming H-bonds or
any other type of specific bond is a random coil. Polyethylene is a simple example. The random
coil model is a helpful starting point for estimating the orders of magnitude of the hydrodynamic
properties of polymers and denatured proteins in solution.
The simplest model of a random coil is a freely jointed chain, in which any bond is free to make
any angle with respect to the preceding one (fig 4.3).
We assume that the residues occupy zero volume, so different parts of the chain can occupy the
same region in space (in reality this one of the main constraints in folding a protein). The model
is obviously a gross oversimplification because a bond is actually constrained to a cone of angles
around a direction defined by its neighbor(fig. 4.4). In a hypothetical 1-dimensional freely-
jointed chain all residues lie in a straight line, and the angle between neighbors is either 0
o
or
180
o
. The residues in a 3-D freely-jointed chain are not restricted to lie in a line or in a plane.
Fig.4.2 Several subunits with specific
tertiary structures pack together,
providing an example of quarternary
structure
Fig 4.3 Freely jointed chain (left) is like a random walk, each step being in arbitrary
direction but of the same length. A better description is obtained by fixing the bond
angle, for example the tetrahedral angle and allowing free rotation about a bond
direction (right)
- 59 -
(a) Measure of sizes.
Consider a 1-D freely-jointed polymer. We can specify the conformation of a molecule by
stating the number of bonds pointing to the right (N
R
) and the number of bonds pointing to the
left (N
L
). The distance between the two ends of the chain is (N
R
-N
L
)l, where l is the length of an
individual bond. We write n=N
R
-N
L
and the total number of bonds N=N
R
+N
L
. The number of
ways of forming a chain with a given end-to-end distance nl is given by the binomial coefficient:
( ) ( ) !
2
1
!
2
1
!
! !
!
)
`
)
`
+
= =
n N n N
N
N N
N
W
R L
(4.1)
The probability that the separation is nl is:
( ) ( )
N
n N n N
N
P
2 !
2
1
!
2
1
!
)
`
)
`
+
= (4.2)
When the chain is compact, meaning that N n << it is more convenient to evaluate P ln : the
factorials are then large and we can use Stirlings approximation in the form:
( ) x x x x |
.
|
\
|
+ + = ln
2
1
2 ln ! ln
2 1
t (4.3)
Then the final result (after some algebra) is:
( ) ( ) ( ) ( ) v v
t
+ + +
|
.
|
\
|
= 1 ln 1
2
1
1 ln 1
2
1 2
ln ln
2
1
n N n N
N
P (4.4)
where N n = v . For a compact coil we use the approximation ( )
2
2
1
1 ln v v v ~ and so
obtain:
2
2 1
2
1 2
ln ln v
t
N
N
P |
.
|
\
|
~ , which rearranges into
N n
e
N
P
2
2 1
2
2
|
.
|
\
|
=
t
(4.5)
This function is plotted in fig 4.5 and can be used to calculate the probability that the ends of a
3-D freely jointed chain lie in the range R to R+dR. We write this probability as fdR, where
2 2
2
3
2
1
4
R a
e R
a
f
|
|
|
.
|
\
|
=
t
t
with
2 1
2
2
3
|
.
|
\
|
=
Nl
a (4.6)
- 60 -
In some coils the ends may be far apart, whereas in others their separation is small. An
alternative explanation of eqn 4.6 is to regard each coil in a sample as ceaselessly writhing from
one conformation to another; then fdR is the probability that at any instant the chain will be
found with the separation of its ends between R and R+dR.
There are several measures of the geometrical size of a random coil. The contour length, R
C
, is
the length of the macromolecule measured along its backbone from atom to atom. For a polymer
of N monomer units each of length l, the contour length is Nl R
c
= . The root mean square
separation, R
rms
, is a measure of the average separation of the ends of a random coil: it is the
square root of the mean value of R
2
, calculated by weighing each possible value of R
2
with the
probability that R occurs;
l N fdR R R
rms
2 1
2 1
0
2
=
|
|
.
|
\
|
=
}
(4.7)
We see that, as the number of monomer units increases, the rms separation of its ends increases
as
2 1
N , and consequently its volume increases as
2 3
N .
Another convenient measure is the size is the radius of gyration, R
g
, the radius of a thin hollow
spherical shell of the same mass and moment of inertia as the macromolecule. It is calculated
from the expression:
2 1
2
2
1 1
|
|
.
|
\
|
=
ij
ij g
R
N
R
(4.8)
The radius of gyration may also be calculated for other geometries. For example, a solid uniform
sphere of radius r has R R
g
2 1
5
3
|
.
|
\
|
= and a long thin uniform rod of length l has ( ) 2 3
2 1
l R
g
=
for rotation about an axis perpendicular to the long axis.
(b) Conformational entropy.
The random coil is the least structured conformation of a polymer chain and corresponds to the
state of greatest entropy. Any stretching of the coil introduces order and reduces the entropy.
Conversely, the formation of a random coil from a more extended form is a spontaneous process
(provided that enthalpy contributions do not interfere).
The conformational entropy of a chain is W k S ln = , where W is given by eq. 4.5. Therefore,
( ) | | !
2
1
ln !
2
1
ln ! ln
)
`
)
`
+ = n N n N N k S
(4.9)
Because the factorials are large (except for large extensions) we can use Stirlings approximation
to obtain:
( ) ( ) ( )
( ) ( ) { }
1 1
2
1
2 1
ln
2
1
ln 2 ln 1 2 ln
+ + +
+
+ + + + =
n N n N
n N n N
N N N k S t
(4.10)
- 61 -
The most probable conformation of the chain is the one with the ends close together (n=0),
which may be confirmed by differentiation. Therefore the maximum entropy is
( ) ( ) N N k S ln 2 ln 1 2 ln
2
1
2 1
+ + = t
(4.11)
The change in entropy when the chain is stretched or compressed by nl is then the difference
between the two corresponding quantities, given by:
( ) ( ) { }
v v
v v
=
+ = A
1 1
1 1 ln
2
1
kN S with N n = v (4.12)
This function is plotted in fig 4.4 and we see that the minimum extension correspond to
maximum entropy.
(c) Constrained chains
The freely jointed chain model can be improved by removing the freedom of bond angles to take
any value. For long chains we can simply take groups of neighboring bonds and consider the
direction of their resultant. Although each successive individual bond is constrained to a single
cone of angle t relative to its neighbor, the resultant of several bonds will lie in a random
direction. By concentrating on such groups rather than individuals, it turns out that for long
chains the expression for the root mean square separation and the radius of gyration should be
multiplied by
Fig. 4.4 the change in molar entropy of a
perfect elastomer as its extension
changes: v-=1 corresponds to complete
extension; v=0, the conformation of
highest entropy corresponds to the
random coil
- 62 -
2 1
cos 1
cos 1
|
.
|
\
|
+
=
u
u
F (4.13)
For tetrahedral bonds, for which
3
1
cos = u (that is 5 . 109 = u
o
),
2 1
2 = F . Therefore:
( ) l N R
rms
2 1
2 = (4.14)
and l
N
R
g
2 1
3
|
.
|
\
|
= . (4.15)
The model of a randomly coiled molecule is still an approximation, even after the bond angles
have been restricted, because it does not take into account the impossibility of two or more atoms
occupying the same space. Such self-avoidance tends to swell the coil, so (in the absence of
solvent effects) it is better to regard R
rms
and R
g
as lower bounds to their actual values.
Fig 4.5 A random coil in three dimensions.
This one contains about 200 units. The root
mean square between the ends (R
rm
s) and
the radius of gyration (R
g
) are indicated.
- 63 -
4.3 The Structure of Proteins
Proteins are the hydrogen atoms of life. (Un-)Fortunately there are many of them, all of them
have a specific function and as a consequence we have to figure out the rules by generalizing
their physical (and chemical and biological) properties. A protein is a polypeptide composed of
covalently linked o-amino acids, NH
2
CHRCOOH, where R is one of only twenty possible
groups. The resulting sequence of R groups linked by peptide bonds for a large part determines
the structure and function of the protein.
4.3.1 The amino acids.
There are 20 different amino acids. Their chemical properties are determined by the R side
groups. These are simple organic molecules composed of C, H, N, O and an occasional S.
Nevertheless, the chemical properties of the various amino acid side chains vary from
hydrophobic to polar to charged, from large to small, from flexible to rigid and these properties
are used to add functionality and activity to a sequence of amino acids folded into a protein
structure. Later we will see a number of examples.
Proteins often contain cofactors for their function. Hemoglobin contains four heme groups
(porphyrin-like structures with an Fe-atom in their center that binds the oxygen molecule),
photosynthetic proteins contain chlorophylls, proteins of the respiratory chain contain hemes,
many protein have binding sites for small ions (K, Ca, Mn), many proteins can be
phosphorylated/dephosphorylated thereby changing their charge etc. etc.
The following figures 4.7 and 4.8 show the 20 amino acids, arranged in four groups: basic,
acidic, non-charged polar, non polar.
Fig 4.6 The condensation of two o- amino acids to form a dipeptide. The
peptide bond is indicated in the red square.
- 64 -
Fig 4.7 The basic side chains. Lysine and arginine are positively charged at pH 7. Note
that the definition of pH inside a protein may be a bit uncertain.
Fig 4.8 The acidic,
uncharged and non-polar
sidechains. The uncharged
polar side chains are often
involved in hydrogen
bonding. The hydrophobic
side chains occur in the
interior of a protein and
their size and shape play an
important role in the
compactness of a protein. In
regions where o-helices
fold over one another small
residues are required.
Proline is special because it
can not fit in the o-helix.
Aromatic residues often
have additional functions.
For instance in photosystem
2 of photosynthesis a
tyrosine plays a crucial role
in electron and proton
transfer. The S atoms in
methionine and cysteine
play important roles in
cofactor binding and
protein folding.
- 65 -
Finally, note that residues will influence each other. In a linear chain the properties of residue i
will be partly determined by residues i-1 and i+1. In a 3-D structure a typical residue sees about
6 others, but it is the precise geometry of the 3-D structure that determines its functionality.
4.3.2 The electronic structure of the peptide bond.
Proteins are sequences of linked o-amino acids, NH2CHRCOOH, where in the living world R is
one of 20 possible groups. For a protein to function correctly, it needs to have a well defined
conformation. For example, an enzyme has its greatest catalytic efficiency only when it is in a
specific conformation. It is the precise sequence of amino acids that determines how a
polypeptide chain is folded and the folded structure is the biologically active molecule. However,
the prediction of the conformation from the primary structure, the so-called protein folding
problem, is extraordinarily difficult and is the focus of much research.
One major factor determining the secondary structure of proteins is found in the stabilization of
certain structures by hydrogen bonds involving the peptide bond. The peptide bond, which is the
unit connecting the C
o
-carbons in a polypeptide chain, is schematically indicated in Fig. 4.6 and
again in 4.9 and 4.10. The peptide bond has some special properties and these properties allow
for the formation of two very stable secondary structures: helices and sheets. For these structures
Pauling and Corey (1951) proposed (without having seen them) that:
(i) The four atoms involved, O, C, N, H lie in a relatively rigid plane. The planarity is
due to the delocalization of t-electrons over the N, C and O atoms and the
maintenance of maximum overlap of the contributing t-orbitals.
(ii) Two types of structures exist, helices and sheets, where all NH and CO groups are
engaged in hydrogen bonding.
(iii) The N, H and O atoms involved in H-bonds between different parts of a polypeptide
chain lie in a (more or less) straight line (with displacements of H tolerated up to not
more than 30
o
from the N-O vector.
The planarity of the peptide bond and its capacity to form H-bonds follows directly from the
electronic structure.
It turned out that when the first crystal structure of a protein (myoglobin) was resolved these
predictions were found to be very accurate.
- 66 -
In the peptide bond the O is sp-hybridized, the C and N atom are sp
2
-hybridized. The bond
angles are shown in Fig. 4.10 and they are close to 120
o
.
The o-framework of the peptide bond is composed of the following bonds: the O-atom has the
configuration ( )
1 1
,
2 2 2
2 2 1
2
O
C O
O
z
sp sp
y
p sp p s o and contributes one electron to a t-
Fig. 4.9 The peptide bond, which is an
essential part of the amino acid chain
constituting a protein, is composed of the
atoms O, C, N, H, which all are positioned in
one plane. Also the two o-carbon atoms
flanking the peptide bond are in that plane.
Consequently the configuration of the
polypeptide backbone is described by two
angles + u, per residue indicated in the
figure.
Fig 4.10 Bond angles in the
peptide bond.
Note that all the angles are
close to 120
o
, typical for sp
2
-
hybridization.
- 67 -
network; the configuration of C is
1 1 1 1 2
2 1
2 2
,
3 2
,
2
,
z
sp sp sp sp sp sp
p s
N C
i
C C O C
o o o
o
, also contributing one t-electron,
and the configuration of N is
2 1
1 ,
1
,
1
,
2
2 1
2 3
1
2 2 2
N
H N
i
C N C N
z
s sp sp sp sp sp
p s o o o
o +
, contributing two t-electrons. A t-
electron network can be constructed by a linear combination of the
N C O
z z z
p p p 2 , 2 , 2 . This will
result in three new t-orbitals to which the four t-electrons can be added (see exercise );
two in the t-orbital with the lowest energy ( | o
t
2
1
+ = E ) and two in the second t-orbital
( o
t
=
2
E ). The resulting energy diagram is shown in Fig. 4.11.
The bonding energy resulting from the t-electrons is sufficient to make the peptide bond rigid
and planar. Note that the t-network does not spread over the
1
,
+ i i
C C
o o
atoms since their 2p
z
-
orbitals are involved in the formation of the sp
3
-hybrids. As a result the peptide chain can twist
around the angles + u, as indicated in Fig. 4.10.
4.3.3 The total energy of a protein and its energy landscape
A polypeptide chain adopts a conformation corresponding to a minimum Gibbs energy, which
depends on its conformational energy, the energy of interaction between different parts of the
chain, and the energy of interaction between the chain and the surrounding solvent molecules. In
the aqueous environment of biological cells the outer surface of a protein is covered by a mobile
sheath of water molecules and its interior may contain pockets of water molecules. These water
molecules play an important role in determining the conformation that the chain adopts through
Fig 4.11 Atomic orbitals, bonding hybrid orbitals and the resulting molecular orbitals of
the atoms C, O and N in the peptide bond.
- 68 -
hydrophobic interactions and hydrogen bonding to amino acids in the chain or to the polypeptide
backbone.
The simplest calculations of the conformational energy of a polypeptide ignore entropy and
solvent effects and concentrate on the total potential energy of all the interactions between non-
bonded atoms. To calculate the energy of a conformation we will use expressions for the non-
covalent interactions described above.
1. Bond stretching. Bonds are not rigid and it may be advantageous for some bonds to
stretch and others to be compressed slightly as parts of the chain press against one another. If we
model a bond as a spring, then the potential energy takes the form of Hookes law and is given
by:
( )
2
2
1
e stretch stretch
R R k V =
(4.16)
where
e
R is the equilibrium bond length and
stretch
k is the force constant, a measure for the
stiffness of the bond in question. Of course this contribution must be summed over all bonds in
the protein.
2. Bond bending. An O-C-H bond angle may open out or close in slightly to enable the
molecule as a whole to better fit together. If the equilibrium bond angle is
e
u we write:
( )
2
2
1
e bend bend
k V u u =
(4.17)
where
bend
k is the bending force constant, a measure for how difficult it is to change the bond
angle. Again this contribution must be summed over all bonds.
3. Bond torsion. There is a barrier to internal rotation of one bond relative to another (just
like the barrier to internal rotation in ethane). Because the planar peptide link is relatively rigid,
the geometry of a polypeptide chain can be specified by the two angles |, that two
neighboring planar peptide links make to each other (see fig.4.9). The sign convention is that a
positive angle means that the front atom must be rotated clockwise to bring it into an eclipsed
position relative to the rear atom. For an all-trans form of the chain all | and are 180
o
. A
helix is obtained when for all links in the chain the| are equal and the are equal. For a right-
handed o-helix
o
57 = | and
o
47 = . For a left-handed helix both angles are positive. The
torsional contribution to the total potential energy is:
( ) ( ) | 3 cos 1 3 cos 1 + + + = B A V
torsion
(4.18)
where A and B are constants of the order of 1 kJmol
-1
. Because for a regular structure, like an o-
helix only two angles are needed to specify the conformation of that helix, and they range from -
180
o
to +180
o
, the torsional potential energy of the entire molecule can be represented on a
Ramachandran plot, a contour diagram in which one axis represents | and the other represents
.
4. Interaction between partial charges. If the partial charges q
i
and q
j
on the atoms i and j
are known, the Coulombic potential energy between them can simply be calculated. Partial
- 69 -
charges for all the atoms in such a structure follow from quantum chemical calculations. Using
the interaction between partial charges does away with the need to take dipole-dipole interactions
into account, for they are taken care of by dealing with each partial charge explicitly.
5. Dispersive and repulsive interactions. The interaction energy of two atoms separated
by a distance r (which we know once | and are specified, assuming that the geometry of each
side chain is known) can be calculated from the Lennard-Jones potential.
6. Hydrogen bonding. In molecular energy and molecular dynamics calculations the
interaction between partial charges (when it includes an H-bond) is fudged to take into account
the effect of H-bonding. In other models H-bonding is added as another interaction (mainly
because of its intrinsic quantum mechanical nature) of the form:
10 12
r
F
r
E
V
bonding H
=
(4.19)
Now the total potential energy of a given conformation ( ) |, can be calculated by summing all
these contributions for all bond angles (including torsional angles) and pairs of atoms in the
polypeptide chain.
Fig. 4.12A shows the potential energy contours for the helical form of polypeptide chains formed
from the non-chiral amino acid glycine (R=H) and the chiral amino acid L-alanine (R=CH
3
), fig
4.13 shows the structure of the o-helix. The contours were computed by summing all the
contributions described above for each choice of angles and then plotting contours of equal
potential energy. Note that the glycine map is symmetrical, with minima of equal depth at
Fig. 4.12 Steric limitations on the bond angles in a polypeptide chain. (A) Each amino acid
contributes three bonds (red) to the backbone of the chain. The peptide bond is planar and does not
permit rotation. By contrast, rotation can occur about the C
o
-C bond ( ) and about the Ca-N
bond (| ). By convention an R group is often used to denote an amino acid side chain (green
circles). (B) the conformation of the main-chain atoms in a protein is determined by the angles
( ) | , for each amino acid. Because of steric collisions between atoms within each amino acid most
pairs of ( ) | , angles can not occur. In this so-called Ramachandran plot each dot represents a
pair of angles actually observed in the structure of a protein.
- 70 -
o o
90 , 80 + = = | and at
o o
90 , 80 = + = | . In contrast, the map for L-alanine is
unsymmetrical and there are three distinct low-energy conformations (marked I, II and III). The
minima of regions I and II lie close to the angles typical of right- and left-handed o-helices, but
the former has a lower minimum, which is consistent with the formation of right-handed helices
from naturally occurring L-amino acids.
A |-sheet is formed by hydrogen bonding between two extended polypeptide chains (large
absolute values of the torsion angles |, . Some of the R-groups point above, some below the
sheet. Two types of structures can be distinguished from the pattern of H-bonding between the
two constituent chains. In an antiparallel |-sheet (see below, fig. 4.20)
o o
113 , 139 + = = |
and the N-H-O atoms of the H-bond form a straight line. This arrangement is a consequence of
the antiparallel arrangement of the chains: every N-H bond on one chain is aligned with a C-O
bond from another chain. Antiparallel |-sheets are very common in proteins. In a parallel |-sheet
(fig 4.23)
o o
113 , 119 + = = | and the N-H-O atoms are not perfectly aligned. This
arrangement is a result of the parallel arrangement of the chains: each N-H bond on one chain is
aligned with a N-H bond of another chain. These structures are not common in proteins.
Although we do not know all the rules that govern protein folding, X-ray diffraction studies of
water-soluble natural proteins and synthetic polypeptides show that some amino acid residues
appear in helical segments more frequently than in sheets, whereas others exhibit the opposite
behavior.
- 71 -
4.3.4 The ohelix and the |-sheet.
Probably the most famous elements of secondary structure in proteins are the o-helix and the |-
sheet. The o-helix was predicted by Linus Pauling (Nobel prize Chemistry in 1954 for his
research into the nature of the chemical bond. Pauling is one of very few people who got the
Nobel prize twice, in 1962 the Nobel peace prize for his initiative to ban the testing of nuclear
weapons, only a few days before his death signed by Albert Einstein).
The right-handed o-helix, characterized by
o
57 = | and
o
47 = and shown in fig. 4.13, is a
very compact structure in which all the peptide links are involved in H-bonding. The diameter of
the helical backbone is about 0.5 nm, one turn of the helix measures 0.54 nm measured along the
long axis of the helix. Helices are extremely popular in compact globular proteins because they
allow the packing of an essentially very polar polypeptide chain into a protein with a very
hydrophobic interior. The first protein for which a 3-D structure was obtained, myoglobin
(Perutz and Kendrew 1956-1961, Nobel Prize Chemistry in 1962) showed 70% of ohelix (see
figs 4.14 and 4.15).
Fig.4.13. The o-helix. A. Polypeptide backbone showing the arrangements of the H-bonds. The N-H
of the peptide bond make an H-bond with the C=O of a peptide bond 3+ a bit residues further along
the chain. And this pattern repeats for every next peptide bond in the chain. B. Ribbon diagram with
the polypeptide backbone drawn in. C. The ribbon symbolizing the o-helix.
- 72 -
Myoglobin binds oxygen via the hemegroup, a Fe-porphyrin. The binding of oxygen can only
occur in a specific geometry, allowing for both strong binding and fast release.
Fig. 4.14 Model of the
myoglobin molecule based
on the 2 X-ray
diffraction map. The white
cord follows the
polypeptide chain. The
model was shown by
Kendrew during his Nobel
lecture.
Fig 4.15 3-D Structure of Myoglobin, a
small oxygen binding protein in muscle. Only
the backbone of protein is indicated plus the
hemegroup. The tube like structures
represent o-helices. Myoglobin consists for
70% of o-helices.
- 73 -
Another important class of proteins which contain a high fraction of o-helix are membrane
proteins. Good examples are the visual protein rhodopsin (see figs 4.17 and 4.18) and the
bacterial photosynthetic reaction center (see fig 4.19). For both crystal structures are available.
The bacterial photosynthetic reaction center was the first membrane protein to be crystallized by
Michel, Deisenhofer and Huber, who got the Nobelprize for Chemistry for this success in 1988.
Fig. 4.16 Fe(II) heme
(ferroprotoporphyrin IX) shown liganded
to a protein His and O
2
as it is in
oxygenated myoglobin and oxygenated
hemoglobin. Note that the heme is a
conjugated system so that all of the Fe-N
bonds are equivalent. Due to the
presence of a second histidine close to
the O
2
molecule, the O
2
must bind at an
angle of about 45
o
, thereby lowering the
binding constant for O
2
, but much more
so for CO.
Fig 4.17 Left: the retina with rods. Right: A rod cell with the folded membrane (red) where
rhodopsin is located
- 74 -
Fig. 4.18. Structure of
Rhodopsin, the visual protein.
Purple cylinders reflect o-
helices, which span the
membrane of a visual cell. The
chromophore, retinal, is
indicated in the center (light
blue). Upon excitation the
retinal chromophore isomerizes
around its C11-C12 double
bond, which leads to a
rearrangement of H-bonds in
the protein structure, eventually
leading to a small
conformational change that
eventually produces a signal to
the brain.
- 75 -
The second most-frequently occurring element of secondary structure is the antiparallel |-sheet.
The N-H and C=O groups of a certain strand are hydrogen bonded to C=O and N-H groups of
adjacent chains that run parallel to it, but in the opposite direction. The R-side groups in each
strand alternately project above and below the plane of the sheet (see fig 4.20)
Fig 4.19. Structure of the bacterial photosynthetic reaction center. The transmembrane part
consists of 11 o-helices: 5 from the L-subunit (yellow), 5 from the M-subunit (red) and 1 from the
H-subunit (green). The blue protein is a 4-heme cytochrome, linked to the reaction center, this
cytochrome subunit is only there in some photosynthetic bacteria. The L and M-subunits are
intrinsic membrane proteins, the H-subunit is mainly in the cytoplasm, the cytochrome is mainly in
the periplasmic space. Upon excitation by a photon an electron is transferred by the
bacteriochlorophyll cofactors from the periplasmic side of the protein to the cytosolic side.
- 76 -
A famous structure in which the |-sheet is the major structural element is the protein fibroin, the
major constituent of silk.
Fig.4.20 The anti-parallel |-sheet. (D) The protein backbone showing the H-bonds
between adjacent strains. (E) Ribbon diagram including the polypeptide backbone
(F) Symbolic representation of the |-sheet.
Fig 4.21 Silk fibroin is organized in an anti-parallel |-sheet.
- 77 -
The cores of many proteins contains extensive regions of |-sheet. These sheets are often used by
proteins to create hydrophobic cavities, well protected from the outside universe and involved in
some kind of special activity. Below we show the structure of the photoactive yellow protein.
Photoactive Yellow Protein; A
bacterial Photoreceptor
Finally, we show two other frequently occurring elements of secondary structure, the parallel
|-sheet and the |-turn.
Fig 4.22 Structure of the Photoactive Yellow Protein, a bacterial photoreceptor,
involved in the detection of harmful blue light. Note the extensive |-sheet structure
flanked by two o-helices that together create a cavity for the light sensor, the p-
coumaric acid chromophore. After absorption of a photon, a structural change is
induced in the protein that triggers the biological response: to swim away from blue
light
- 78 -
|-sheets and |-turns
4.4 Protein Folding.
Proteins fold into a unique 3-D structure with a unique biological function. Even a mutation of
one single amino acid may perturb this process in the sense that no longer a stable structure can
be formed, or if it is formed it has no, or a much reduced, biological activity.
Which interactions determine how a protein folds? Of course the folded state must represent a
minimum in energy, meaning that to solve the problem of protein folding we have to be able to
find the true minimum of the energy of the protein. Here we will give some simple rules.
1. Two atoms can never be in the same place. Steric hindering is one of the major driving
forces for the formation of secondary structure (see fig. 4.24) structure.
Fig 4.23 Parallel |-sheets and |-turns.
- 79 -
2. Covalent connections between different parts of the chain can be made by di-sulfide
bridges, involving two cysteines (see fig 4.25 and 4.26).
Fig. 4.25 Two cysteine residues on
different parts of the polypeptide
chain can be covalently linked via
the reduction of O to form H
2
O. A
disulfide bridge in a protein is
exceptionally stable.
Fig. 4.26 Disulfide bridges in a protein.
When a protein is being produced by the
ribosome, a certain number of cysteine
residues will be present. It is of crucial
importance that the correct disulfide bridges
are formed!
Fig 4.24 Steric interference
between the carbonyl
oxygen and amide hydrogen
in two neighboring peptide
bonds of a polypeptide
chain.
- 80 -
3. Non-covalent interactions.
As a result of ionic interactions, Van der Waals forces and hydrogen bonds, each type of protein
has a particular three dimensional structure, which is determined by the order of the amino acids
in the chain. This is illustrated in Fig.4.27.
The final folded structure, or conformation, adopted by any polypeptide chain is generally the
one in which the free energy is minimized (note that this minimum may not be unique for a
system with so many degrees of freedom). For globular, water soluble proteins this is generally a
compact conformation with a hydrophobic core and polar/charged residues (such as arginine,
glutamine, histidine etc.) exposed to the aqueous phase (see fig 4.28).
Fig. 4.27. Three types of noncovalent bonds that help proteins fold.
- 81 -
When polar residues are buried in the inside of a protein they are generally hydrogen-bonded to
other polar amino acids or to the polypeptide backbone, as illustrated in Fig. 4.29.
Fig. 4.28 How water-soluble protein folds into a compact configuration. The polar amino
acid side chains tend to gather on the outside of the protein, where they can interact with
water; nonpolar amino acid side chains are buried on the inside to form a tightly packed
hydrophobic core of atoms that are hidden from water. In the schematic drawing the
protein is small and contains about 30 amino acids.
Fig 4.29 Hydrogen bonds in a protein. Large number of hydrogen bonds form between adjacent
regions of the folded polypeptide chain and help stabilize its 3-D shape. Three types of H-bonds
are indicated.
- 82 -
Protein folding has been studied in a test tube by using highly purified proteins. A protein can be
unfolded, or denatured, by treatment with certain solvents, which disrupt the noncovalent
interactions holding the folded protein chain together. This treatment converts the protein into a
flexible polypeptide chain that has lost its natural shape. When the denaturing solvent is
removed, the protein often refolds spontaneously, or renatures, into its original conformation (as
is illustrated in Fig. 4.30), indicating that all the information needed for specifying the three-
dimensional shape of a protein is contained in its amino acid sequence.
Each protein normally folds up into a single stable configuration. However, this conformation
often changes slightly when the protein interacts with other proteins in the cell. It is this
modulation of the shape of proteins by its interaction with the environment that is often crucial
to the function of the protein.
Although a protein chain can fold into its correct conformation without outside help, protein
folding in a living cell is often assisted by special proteins called molecular chaperones. These
proteins bind to partly folded polypeptide chains and help them progress along the energetically
most favorable folding pathway. Chaperones are vital in the crowded conditions of the
cytoplasm, since they prevent the temporarily exposed hydrophobic regions in newly synthesized
proteins from associating with each other to form protein aggregates. However, the final three-
dimensional shape of a protein is still dictated by its amino acid sequence: chaperones simply
make the folding process more reliable.
To understand how a protein folds into its functional state is a formidable problem. Take a
simple protein of 100 amino acids, say for every amino acid there are 10 possible conformers,
implying that there are 10
100
possible structures to be investigated during the folding process.
Suppose it takes 10
-10
s to test a certain structure then it would take > 10
80
years to look at all of
them. So protein folding can not be a random search through conformational space.
Fig. 4.30. The refolding of a denatured protein. This experiment demonstrates that the
conformation of a protein is determined solely by its amino acid sequence.
- 83 -
A distribution of unfolded species slide on the same
energy surface to reach the minimal free energy
They meet at the saddle point where key residues
have formed their native like contact.
The fact that the correct folding of a protein requires some unstable intermediate has the
consequence that there is a finite probability that the intermediate misfolds or aggregates. There
are two ways to aggregate: in some random way (like your boiled egg on sunday morning) or in
some organized way, like in a fiber. The latter aggregates may be extremely stable and are
associated with certain diseases. Thus if the subtle balance in this process is perturbed these
highly organized and stable aggregates will accumulate.
Fig 4.31 Free energy of a folding protein plotted as a function of the number of
contacts between residues (not all are favorable) and the number of native
contacts (meaning contacts that also occur in the functional protein). The
potential energy drives the system to a conformation where a certain number of
native contacts has been established, but the chain is not yet folded. Note that
there are many possible pathways. Once this point is reached, the chain folds
rapidly.
Question: What would the potential energy landscape look like if a certain fraction
of the paths would very quickly lead to the formation of an o-helix? Would that help
protein folding? Explain
- 84 -
In vitro, X-ray
Highly
organised
misfolded
structure
.Disease
Parkinsons
disease
Fig. 4.32. A unified view of the types of structures that can be formed by
polypeptide chains.
Fig 4.33 Some structures observed as a consequence of protein misfolding
- 85 -
4.5 Respiration
Also non-photosynthetic cells need energy. They burn high energy compounds (food) to
generate the energy they need via a process called respiration. In eukaryotic cells respiration
occurs in the mitochondrion. Interestingly, mitochondria (like chloroplasts) carry their own
DNA, suggesting a complex evolutionary origin.
In essence the process of respiration proceeds as follows: food is chemically modified in reduced
compounds (NADPH, sugars) which serve as an electron donor. The respiratory chain consists of
a set of membrane bound proteins, via which the electron is transported to reduce oxygen (that
we breathe) to water. During this electron transport reaction protons are transported across the
mitochondrial membrane to build up a proton gradient. The proton gradient is used by the
enzyme ATPsynthase to produce ATP, the universal energy compound of the living cell.
Fig 4.35 Harnessing energy for life. (A). The essential requirements for chemiosmosis are a
membrane in which are embedded a pump protein and an ATP synthase plus a source of high-
energy electrons. The protons (H+) shown are freely available from water molecules. The pump
harnesses the energy of electron transfer to pump protons across the membrane, creating a proton
gradient. (B) This proton gradient serves as an energy storage that can be used to drive ATP-
synthesis by the ATP-synthase enzyme. The red arrow shows the direction of proton movement at
each stage
- 86 -
The bc1 complex is one of the components of the respiratory chain. It is a trans-membrane
protein, meaning that the protein spans a very hydrophobic membrane between two aqueous
phases. The bc1 complex oxidizes UQH
2
, the two protons are delivered to the periplasmic side of
the membrane (while they were originally taken up by UQ at the cytoplasmic side), thus building
up a proton gradient. One of the two electrons is transported to O
2
to be reduced to H
2
O. The
other is transferred once more across the membrane via an electron transfer chain to be used to
put another proton across the membrane.
In living cells this trans-membrane electrochemical potential may easily be of the order of a few
100 mV, corresponding to an electrical field of about 10
6
V/cm, even not easy to achieve under
laboratory conditions!!.
Fig 4.36 The general mechanism of oxidative phosphorylation. As a high energy electron is
passed along the electron transport chain, some of the energy that is released, is used to
drive the three respiratory enzyme complexes that pump H
+
out of the matrix. The resulting
electro-chemical proton gradient across the inner membrane drives H
+
back through the
ATP-synthase, a membrane protein complex that uses the energy of the H
+
flow to
synthesize ATP from ADP and Pi in the matrix.
- 87 -
The H
+
-gradient generated by the transport of electrons from NADH to O
2
down the respiratory
chain is used to convert ADP plus inorganic phosphate P
i
into ATP, the energy carrier of the
living cell. The protein involved, the ATP synthase binds in a first step ADP and Pi, the
backflow of protons leads to a conformational changes that favors the formation of ATP,
subsequently the ATP is released. This 3-step process is coupled to a rotation of the enzyme
around an axis perpendicular to the membrane which has been visualized using microscopy by
binding a long actin polymer, labeled with fluorescent dyes to the rotating enzyme.
membrane
Fig 4.37 The bc1 complex, one of the proteins of the respiratory chain. The complex accepts
electrons from reduced ubiquinone (UQH
2
, ubiquinol, doubly reduced, doubly protonated
ubiquinone) produced from food. The two protons are put into the periplasmic volume, one electron
reduces cytochrome c, a small water soluble protein that delivers the electron via the cytochrome
oxidase to O
2
to form water, the other electron is transported to the other side of the membrane via
two cytochrome b hemes, attached to the bc1 complex to repeat the same trick again together with
the electron from another UQH
2
. In this way the energy of food is optimally converted into a proton
gradient.
UQH
2
->UQ
Cyt c
2H
+
e
-
e
-
e
-
e
-
o-helix
Cyt b
UQ
H
+
O
2
periplasm
cytoplasm
Cyt b
2H
+
UQ->UQH
2
2e
-
- 88 -
ATP-synthase
(A) The enzyme is composed of a head portion called the F1 ATPase and a transmembrane H+
carrier, called F0. Both F1 and F0 are formed from multiple subunits, as indicated. A rotating
stalk rurns with a rotor formed by a ring of 10-14 c subunits in the membrane (red). The stator
(green) is formed from transmembrane a subunits, tied to other subunits that create an elngated
arm. The arm fixes the stator to a ring of 3o and 3| subunits that form the head. (B). 3-D
structure of the F1 ATPase determined by X-ray crystallography. (Abrahams et al., Nature 370:621-628)
Fig. 4.38 Summary of the ATP-synthase structure and mode of operation While
protons move down a proton gradient, they induce the rotation of the F0 part of the
complex, which is coupled to a long shaft that then rotates in the core of the F1
subunit. Since this shaft is not cylindrically symmetric, its motion causes rotating
conformational changes in each o|-heterodimer (see 4.11A)
Fig. 4.39 The three stages of the ATP-
synthase as a function of the position of the
rotating shaft. In L Pi and ADP are bound,
in T the equilibrium is shifted to form ATP,
in O the ATP is released. Note that it is NOT
the complex that rotates, it is the chemical
status of each of the pair of subunits that
rotates as a consequence of the rotation of
the shaft in the center.
- 89 -
4.6 Membranes
All cells are enclosed in a plasma membrane. This container acts as a selective barrier that enables
the cell to concentrate nutrients gathered from its environment and retain the products it has
synthesized for its own use, while excreting waste products. Without its plasma membrane the cell
could not maintain its integrity as a coordinated chemical system. This membrane is formed by a set
of molecules, that have the simple physico-chemical property of being amphipathic, that is, they
consist of one part that is hydrophobic (dislikes water) and another part that is hydrophilic (likes
water). When such molecules are placed in water they aggregate spontaneously arranging their
hydrophobic portions to be as much in contact with one another as possible to hide them from the
water, while keeping their hydrophilic parts exposed. Amphipathic molecules of appropriate shape,
such as the phospholipid molecules that comprise most of the plasma membrane, spontaneously
aggregate in water to form a bilayer that creates small closed vesicles. This can be easily
demonstrated in a test tube. Note that now the aqueous content of the vesicle is isolated from the
external medium. Although the chemical details vary, the hydrophobic tails of the predominant
membrane molecules in all cells are hydrocarbon polymers (-CH2-CH2-CH2-....) and their
spontaneous assembly into a bilayered vesicle is but one of many examples of an important general
principle of life: cells produce molecules whose chemical properties and 3-dimensional structure
cause them to self-assemble into the functional units that the cell needs.
Fig 4.40 Cartoon of the plasma membrane. The plasma membrane separates the cytosol
from the extracellular space. Integral membrane proteins, like receptors and channels,
cross the membrane. Receptors often have specific oligosaccharides attached to them that
play a role in the recognition of specific compounds like hormones. Binding of a hormone
then leads to a conformational change that is sensed at the cytosolic side of the membrane.
Channels are proteins that selectively transport certain compounds across the membrane
(water, ions, sugars, other proteins), sometimes passively, sometimes by using the
electrochemical gradient that is maintained across the membrane. Peripheral membrane
proteins are attached to the plasma membrane, they do not cross the membrane.
- 90 -
4.6.1 Formation of the lipid bilayer
Phospholipids are the amphiphilic molecules that form biological membranes. They consist of a
water-loving head group and a water-avoiding hydrocarbon tail. The water-avoiding tails tend to
cluster together. Immersed in water they spontaneously organize themselves to form a lipid bilayer,
with the hydrophilic headgroups exposed to the water and the hydrophobic tails forming a
hydrophobic core of bilayer (see fig 4.41).
Fig 4.42 shows some of the most frequently occurring lipids, in total there are few hundred known.
The table below summarizes the permeability coefficients for some ions and molecules for a
synthetic and for a biological membrane
Fig 4.41 Formation of a membrane by amphipathic
phospholipid molecules. These molecules have a
hydrophilic (water-loving, phosphate) head group and a
hydrophobic (water avoiding) hydrocarbon tail. At the
interface of oil and water they arrange themselves as a
single sheet with their headgroups facing the water and
their tails facing the oil. When immersed in water they
arrange themselves in a phospholipids bilayer enclosing
an aqueous compartment
- 91 -
Biological membranes are highly impermeable to water and ions. As a consequence specific proteins
exist that transfer ions, water, sugars etc across the membrane.
4.6.2 Transport across a membrane
A multitude of compounds must be transported into and out of the cell via the plasma membrane,
into and out of the nucleus, mitchondria, chloroplasts, the golgi apparatus etc, including
synthesized proteins!!!
Fig. 4.42 Structures of Phosphatidylethanolamine, Phosphatidylserine and Phosphatidylcholine.
Functional groups derived from esterified alcohols are shown in blue. Since each of these lipids can
contain many combinations of fatty acyl groups, the general name refers to a family, not to a single
molecule.
- 92 -
Fig 4.43 Membrane transport proteins. (A) Structure of a molecule of bacteriorhodopsin,
from the archaebacterium Halobacterium halobium. This transport protein uses the energy
of absorbed light to pump protons (H
+
) out of the cell and form a proton gradient. The
polypeptide chain threads across the membrane; in several regions it is twisted into an o-
helical conformation and the helical segments are arranged such to form the walls of a
channel through which the H
+
ions are transported. (B) Diagram of the set of transport
proteins found in the membrane of the bacterium Thermotoga maritima. The numbers in
parentheses refer to the number of different membrane transport proteins that occur of each
type. Most of the proteins within each class are evolutionarily related to one another and to
their counterparts in other species
- 93 -
MscL - Structure
Transmembrane proteine
Channel proteine forms a large
non-selective safety valve to
protect the cell from lysis by
osmotic downshocks
Abundance: Plants, bacteria,
fungi, cardiovascular regulation
in animals (eg. kidneys)
Importance: Highly convenient
molecular system for studies of
elemental principles of
mechanotransduction
Periplasmic side
Cytoplasmic side
Crystal Structure: Chang et al Science, 282 (1998), 2220.
Postulated helical structure of S1, also: Sukharev et al., Biophys. J., 81 (2001), 917.
Tb-MscL closed (relaxed) form
Crystal structure: 3.5 resolution
Porines are trans-membrane proteins involved in transport. They are barrel-like structures made
out of a variety of |-strands.
Porine
Fig. 4.44 Structure of the mechanosensitive sensitive channel MscL. The channel
opens in response to pressure and functions as a safety valve to protect the cell
from lysis by osmotic pressure.
Fig. 4.45
- 94 -
Specific ion pumps are able to exchange ions against a concentration gradient using ATP.
[Na
+
]=140 mM
[Na
+
]=10 mM
[K
+
]=100 mM
[K
+
]=5 mM
A+=70 mV
3 Na
+
2 K
+
1 ATP
Here we consider the energetics of the K
+
-Na
+
exchange. The free energy change for the
transport of an ion across the membrane is given by:
A + = A ZF
C
C
RT G
out
in
ln
F is faraday constant (96.5 kJ mol
-1
V
-1
). A+ is the membrane
potential in volts and measured from out->in. Thus with a positive
membrane potential the transport of cations into the cell is favoured
mol kJ
mol
V
J
K mol
K
J
G
/ 55 . 13
07 . 0 . 48 . 96 1
10
140
ln 310 . 3 . 8
=
+ = A
Thus per mole of Na
+
For 3 moles we have 40.65 kJ/mol
Idem for 2 moles of K
+
from outside to inside: AG=1.94 kJ/mol
Hydrolysis of 1 mole of ATP under standard conditions yields
-30 kJ/mole. However, in the cell the concentrations of P
i
and ADP
are relatively high.
Fig. 4.24. An ATP-driven Ca-pump
Fig 4.46 The ATP-driven K
+
-Na
+
exchanger. Na
+
and K
+
are exchanged and
transported across the membrane against their gradients using ATP.
- 95 -
5. The photosynthetic pigments
The chlorophylls play a major role in the process of light absorption, excitation energy transfer
in the light harvesting antenna and charge separation in the reaction center. In general they form
complexes with proteins ( pigment-protein complexes), with the chlorosomes of the green photosynthetic
bacteria containing large BChl c aggregates being an exception. We will discuss the
intrinsic spectral and energetic properties of the chlorophylls, but we emphasize that the in vivo
environment of these pigments has a major impact on them. The other group of pigments that we
will discuss are the carotenoids. They are largely responsible for the colors in nature. In natural photosynthesis
carotenoids fulfill many roles. Firstly they are active in light-harvesting by absorbing
photons in the 400-500 nm region and transferring the energy to (bacterio-)chlorophyll. In fact
the energy transfer from spirilloxanthin to bacteriochlorophyll in the LH1 antenna of the purple photosynthetic
bacterium Rhodospirillum rubrum was the first excitation energy transfer process
to be quantitatively measured by Duysens in the 1950s. Their second, and possibly most important,
role is in photo protection by removing harmful triplets from chlorophyll, that otherwise would react
with oxygen to produce singlet oxygen. Since the formation of singlet oxygen must be prevented at
all costs, the chlorophyll triplet quenching has to be very efficient, requiring close contacts between
chlorophylls and carotenoids. Consequently, chlorophylls and carotenoids always occur in a specific structure
imposed by the protein environment. Thirdly, carotenoids have a structural role.
In Lhc2 (the major plant light harvesting complex) the two central luteins form an essential
element in the structure of the whole complex.
Finally, carotenoids have been implicated in antenna regulatory processes, allowing the
light-harvesting antenna to switch from a state in which it can function as an energy donor to a
state where it may act as an energy sink. How this switching takes place is unknown.
Pigments 1. The Chlorophylls.
The chemical formula for chlorophyll is C
55
H
72
N
4
O
5
Mg. This simple representation is entirely inadequate
to convey the essential properties of this extraordinary molecule. The structural formula for chlorophyll a
is shown in fig.5.1. It is a squarish planar molecule, about 1 nm on a side. A Mg atom in the center of
the planar part is coordinated to four N-atoms. These nitrogens are each part of a sub structural element
of the molecule that is derived from pyrrole, a cyclic compound with a nitrogen atom in a five-membered ring
with four carbons. For this reason chlorophylls and related compounds are often referred as tetrapyrroles. A
fifth ring is formed in the lower right corner, and a long hydrocarbon tail is attached
to the lower left (in the standard representation). Chemically, the chlorophylls are related to the
porphyrins (heme), which are also tetrapyrroles, but porphyrins are generally more symmetric.
- 96 -
The five rings in chlorophylls are lettered A through E, and the substituent positions on the macro cycle
are numbered clockwise, beginning in ring A, as shown in fig5.1 according to the officially
recognized International Union of Pure and Applied Chemistry (IUPAC) nomenclature. An older nomenclature
known as the Fischer system (and often used in old and even modern photosynthesis literature) is also shown.
By convention, the y molecular axis of all chlorophylls is defined as passing through the N atoms of
ring A and C, with the x-axis passing through the N atoms in rings B and D. The z-axis is perpendicular
to the plane of the plane of the macro cycle. A delocalized t electron system extends over
most of the molecule, with the exception of ring D, in which the C-17-C-18 double bond is reduced to a
single bond. The tail is formed by condensation of four isoprene units and is then esterified to ring D.
It is often called the phytol tail, after the polyisoprenoid alcohol precursor that is attached during biosynthesis.
It is also sometimes called the isoprenoid tail.
Most of the chlorophylls are classified as chlorines rather than porphyrins, by virtue of the reduced
ring D. Most of the bacteriochlorophylls are similarly called bacteriochlorins, because of the reduction
of rings B and D. All chlorophylls and bacteriochlorophylls contain the extra ring E, which is called
the isocyclic ring.
Most chlorophyll-type pigments contain three chiral atoms, C-13
2
, C-17 and C-18. Bacteriochlorophyll
contains two additional chiral centers, C-7 and C-8. In all cases, the stereochemical fidelity of the
biosynthetic enzymes is extremely high, so the compounds found in cells are a single species and not mixtures.
Fig 5.1. Numbering schemes for chlorophylls and bacteriochlorophylls. Chlorophyll a is
shown, although the same basic numbering scheme applies to all chlorophyll-type
pigments. Left, the current IUPAC standard system. Right, the older Fischer numbering
system. Asterisk indicate chiral carbon atoms
- 97 -
The structures of bacteriochlorophyll a and b are shown in fig5.2.
Fig5.2. The structure of bacteriochlorophyll a (left) and bacteriochlorophyll b (right). Note that
in bacteriochlorophyll in addition to ring D also ring B has been reduced. Bacteriochlorophyll a
occurs in many photosynthetic purple bacteria; bacteriochlorophyll b for instance in the purple
bacterium Rhodopseudomonas viridis.
Chlorophyll a
Chlorophyll a, probably the most abundant large organic molecule on earth, is found in all known eukaryotic
photosynthetic organisms. Among prokaryotes, it is found in large quantities only in the cyanobacteria
(including the prochlorophytes), although traces of minor variants of chlorophyll a
are found in some anoxygenic bacteria, the green sulfur bacteria and heliobacteria, where it has an
important function as an intermediate in the electron transport chain. An important variant of
chlorophyll a is chlorophyll a. This pigment differs from chlorophyll a only in the stereochemistry
at the C-13
2
position. It is found in small but reproducible amounts in photosystem I complexes of
plants, where one molecule forms half of P700, the special pair of pigments that is the primary
electron donor (see below). The spectral and redox properties of chlorophyll a are very similar to
those of chlorophyll a.
Chlorophyll b
Chlorophyll b is identical to chlorophyll a except at the C-7 position where a formyl
group (-COH) replaces the methyl group (-CH
3
). This change shifts the absorption maximum
about 20 nm to the blue (650 nm for chlorophyll b vs. 670-680 nm for chlorophyll a). Chlorophyll b
is the major accessory light-absorbing pigment in the majority of eukaryotic photosynthetic organisms,
with the exception of the red an brown algae. In Lhc2, the peripheral light-harvesting complex of photosystem
2, and the most abundant protein on earth, there are 6 chlorophylls b and 8 chlorophylls a. Following light
absorption the chlorophylls b transfer their electronic excitation to chlorophyll a
within a few picoseconds (see below). In photosynthetic prokaryotes it is found only in the
prochlorophytes.
- 98 -
Chlorophyll c
Chlorophyll c is more a porphyrin and not a chlorin, since it does not have ring D reduced and
it does not have a phytol tail. As a consequence its Q
y
extinction coefficient is weak. Chlorophyll c
is found exclusively in various marine algae like diatoms and dinoflagellates. It functions in Lhc2-like antenna
complexes and transfers energy to chlorophyll a.
Chlorophyll d
Chlorophyll d is different from chlorophyll a in only one regard: the substituent at the C-3 position
is a formyl group in chlorophyll d, instead of the vinyl group found in chlorophyll a. Until recently, chlorophyll
d was known only as a trace constituent of certain algae and was suspected to be an
artifact. However, in 1996 a cyanobacterium, Acaryochloris marina, was discovered as a symbiont
in a marine invertebrate. This organism contains chlorophyll d as its major pigment, although it
also contains chlorophyll a and bilin pigments. Chlorophyll d is energetically and structurally an intermediate
between chlorophyll a and bacteriochlorophyll a. It has a red-shifted (about 80 nm)
absorption (so drives oxygenic photosynthesis with relatively low energy photons!!!). The discovery
of this pigment in a living organism has prompted suggestions that it, or a related pigment, may
have been an intermediate in the evolutionary transition from anoxygenic to oxygenic photosynthesis.
Bacteriochlorophyll a
The chemical structure of bacteriochlorophyll a is shown in fig 5.2. It is the principal chlorophyll-like pigment
in the majority of anoxygenic photosynthetic bacteria. The chemical differences between the structures of
chlorophyll a and bacteriochlorophyll a are the acetyl group at the C-3 position and the single bond between C-
7 and C-8, instead of the double bond found in chlorophylls. This reduces the degree of conjugation in the
macrocycle and thus reduces the symmetry of the molecule compared with chlorophylls. These structural
changes exert remarkable and major effects on the spectral properties, which will be discussed below.
A few species of the purple photosynthetic bacteria have been found that use Zn as the central metal ion instead
of Mg. These organisms are found in highly acidic environments where Mg
2+
is readily displaced by H
+
. Zn
and Mg are the only metal ions that have been found incorporated in natural chlorophylls, although many other
metals can be inserted synthetically into the metal-free pigments. The reason for this specificity is probably that
Mg is very readily available, whereas Zn is a trace element in almost all environments and would therefore
often be a limiting nutrient. As discussed below, most other metals are unsuitable for photosynthesis, because
pigments with these metals incorporated exhibit very short excited state lifetimes.
Bacteriochlorophyll b
Bacteriochlorophyll b is found only in a few species of purple bacteria. It differs from bacteriochlorophyll a
only by the presence of an exocyclic double bond in ring B. Its chemical structure is shown in fig 5.2.
Bacteriochlorophyll b is special because it has the longest-wavelength absorbance band of any known
- 99 -
chlorophyll-type pigment. In vivo, in a purple bacterium called Rhodopseudomonas viridis the absorption max
is in the region 950-1050 nm.
Bacteriochlorophyll c, d, e
These bacteriochlorophylls will be considered as a group, because they are found only in green photosynthetic
bacteria, organisms that contain a special antenna complex known as the chlorosome. In BChl c Ring B
contains a C-7-C-8 double bond, as in chlorophyll, making these pigments chlorines instead of
bacteriochlorins. They also have a hydroxyethyl substituent at the C-13
1
position in ring E. This functional
group is essential to the aggregation of these pigments in the chlorosome. These bacteriochlorophylls show a
natural tendency to form large aggregates (10000 of Bacteriochlorophylls) which form highly organized helical
structures that absorb light very efficiently and show ultrafast energy transfer to some membrane bound energy
acceptor. In the green bacterium these aggregates are surrounded by a membrane and the whole complex
associates to the photosynthetic membrane.
Pheophytins and bacteriopheophytins
The metal-free chlorophylls are known as pheophytins. The structures of pheophytin a and of
bacteriopheophytin a are shown in fig. 5.3
5.2 Spectroscopic Properties of Chlorophylls
The chlorophylls all display two major absorption bands, one in the blue or near UV region and one in the red
or near IR region. The lack of significant absorption in the green gives chlorophylls their characteristic green or
blue-green color. These absorption bands arise from t->t* transitions, involving the electrons in the
conjugated t system of the chlorin macrocycle. The absorption spectra of chlorophyll a and
bacteriochlorophyll a are shown in figs 5.4 and 5.5.
Fig 5.3 Chemical structures of pheophytin a and bacteriopheophytin a.
Note the presence of the two protons in the heart of the pheophytins.
Pheophytins are essential components of plant and bacterial reaction
centers, most likely because they are relatively easy to reduce
- 100 -
Fig. 5.4 Absorption (left) and fluorescence (right) spectra of chlorophyll a in diethyl
ether. The extinction coefficient of chlorophyll a at 670 nm is about 100 mM
-1
cm
-1
. Next to
the Q
y
(polarized along the y-axis) transition in the red and the Soret transitions in the
blue, the weak Q
x
(polarized along the x-axis) is indicated.
Fig 5.5 Absorption (left) and fluorescence (right) spectra of bacteriochlorophyll a in diethyl
ether. Note the 100 nm redshift of the spectrum and the enhanced splitting between the Q
y
and
Q
x
transitions. Both are a consequence of the further loss of symmetry in bacteriochlorophyll a
as compared to chlorophyll a. The extinction coefficient of bacteriochlorophyll a around 770
nm is about 100 mM
-1
cm
-1
- 101 -
These spectra can be described theoretically by using a four orbital model originally proposed by Gouterman.
The four t MOs that are principally involved in these transitions are the two highest occupied MOs and the
two lowest unoccupied MOs. The MO energy diagram and the transitions predicted for a symmetric
porphyrin, chlorin and bacteriochlorin are shown in fig 5.6. The two lowest transitions are called Q, and the
two higher-energy ones are know as the B- or Soret bands
The diagram shown in fig 5.6 is an oversimplification of what is a complex relationship between
electronic states and orbital energies. It suggests that electronic transitions reflect a simple promotion of
an electron from a HOMO to a LUMO. In reality, several different electronic configurations, including
contributions from much higher-energy MOs, can contribute to the electronic transition. This
phenomenon is known as configuration interaction. The result is that there is not a simple one-to-one
correspondence between orbital occupations and electronic transitions. However, it is also the case that
Fig. 5.6 MO energy level diagram of porphyrin, chlorin and bacteriochlorin. A very
simplified representation of the electronic transitions is indicated
- 102 -
the majority of contributions to the transition can come from a single configuration. So the picture in
fig. 5.6 indicates the relative changes in orbital energies and transition energies as one goes from the
more symmetric porphyrin to the very asymmetric bacteriochlorin.
The electronic transitions are associated to transition dipole moments (see chapter 6) with different
strengths and orientations. The longest-wavelength transition is invariably polarized along the y-axis of
the molecule and is therefore known as the Q
y
transition. This means that the absorption will be
strongest if the electric field vector of linearly polarized light is parallel to the y molecular axis of the
pigment. The exciting light couples to the t-electrons of the molecule and transiently arranges them
somewhat during the transition. The Q
y
transition causes a shift in electron density that is directed along
the y molecular axis of the molecule as is illustrated in fig 5.7. The weaker Q
x
transition in
bacteriochlorophyll a is also polarized but along the x molecular axis, also shown in fig 5.7. For
chlorophyll a, the Q
y
transition is polarized along the y molecular axis. The Q
x
transition, however, is
not as well resolved as in bacteriochlorophyll and quantum chemical calculations suggest that it is not
polarized directly along the x molecular axis. The soret bands have a mixed polarization.
In addition to the fundamental Q and B electronic transitions, vibrational overtone transitions can also be
observed, especially on the Q
y
band. These represent a simultaneous vibrational and electronic excitation, with
the final state being an excited vibrational state of the excited electronic state. A progression of vibrational
states can be observed, with the most intense transition the 0,0 band, with higher energy satellites termed 0,1;
0,2 etc. The first number is the vibrational number of the ground electronic state before light absorption, and
the second number is the vibrational state of the excited electronic state after the transition. Of course there are
many vibrations in a chlorophyll molecule, but only one is strongly coupled to the optical excitation of
chlorophyll and thus responsible for the vibronic structure in the absorption spectrum (see below)
Fig 5.7 Electronic density changes associated with Q
x
and Q
y
transitions in
bacteriochlorophyll a. Note that the signs are chosen arbitrarily, the transition charge
density changes oscillate with the incident light field.
- 103 -
The fluorescence spectrum of all chlorophylls (and of all big molecules) peaks at slightly longer wavelengths
than the absorption maximum. The fluorescence emission is polarized along the y-molecular axis, as it is
emitted from the lowest excited state corresponding to the Q
y
transition. The fluorescence spectrum generally
displays a characteristic mirror image relationship to the absorption. This is because the ground and excited
state potential energy surfaces have similar shapes, so those molecular vibrations that are activated during
electronic absorption are likely to be activated upon fluorescence emission. However in this case the initial
state is the ground vibrational state of the excited electronic state and the final state is the excited vibrational
state of the ground electronic state. This causes a shift of the emission to longer wavelengths, known as the
Stokesshift. For pigments in a polar solvent (water, protein) there is an additional term to the Stokes which
reflects the reorientation of solvent molecules/amino acids as a response to the changed charge distribution of
the chromophore in the excited state.
5.3 Carotenoids
Carotenoids are found in all known native photosynthetic organisms, as well as in many nonphotosynthetic
organisms (like in the human eye!!!!). There are many hundreds of chemically distinct carotenoids, so it is
impossible to mention them all. However, there are some consistent structural features that are common to
most photosynthetic carotenoids. They are extended molecules with a delocalized t electron system.
Carotenoids from oxygenic organisms usually contain ring structures at each end, and most carotenoids contain
oxygen atoms, usually as part of hydroxyl or epoxide groups. Structures of several of the carotenoids found in
photosynthetic systems are shown in fig. 5.8. Carotenoids are synthesized in such a manner that the number of
conjugated carbon atom in the chain increases. This has the effect of shifting the major absorption bands into
the visible region. The end product is carotenoid lycopene, which is responsible for the red color of tomatoes.
In most organisms there are two additional stages: cyclization of the ends of the molecule (see |-carotene)
followed by derivatization by hydroxylation or any of a wide variation of other processes.
Carotenoids have several well-documented essential functions in photosynthetic systems. First, they are
accessory pigments in the collection of light, absorbing light in the blue-green part of the solar spectrum and
transferring the excited state energy to a closeby chlorophyll. Most antenna complexes contain carotenoids.
Second, carotenoids function in a process called photoprotection. Carotenoids rapidly quench triplet excited
states of chlorophylls before they can react with oxygen to form the highly reactive and damaging excited
singlet state of oxygen (called singlet oxygen). Furthermore, carotenoids also quench the singlet oxygen of it is
somehow formed. Finally carotenoids have been shown to be involved in the regulation of energy transfer in
the antennas. This process, which is connected to the xantophyll cycle, avoids over-excitation of the
photosystem 2 reaction center by allowing the light-harvesting system to switch to a state where excitations are
no longer delivered to the RC, but, instead, are dissipated into heat.
Carotenoids have very unusual energetic and spectroscopic properties. They usually exhibit an intense
absorption in the 400-500 nm range, giving them their characteristic orange color. However it was discovered
that this is a transition from the ground state (S
0
) to the second excited singlet state (S
2
)
instead of to the first excited singlet state (S
1
). The optical transition from S
0
to S
1
is forbidden because
of the symmetry of the carotenoid molecule. An energy level diagram typical of many carotenoids is
shown in fig. 5.9. The lifetime of S
2
is very short, usually relaxing to S
1
by internal conversion on a
subpicosecond (100 fs) timescale. This short lifetime of S
2
means that the fluorescence of the S
2
state is highly
quenched and is not observed in most situations. From S
1
the excited carotenoid can relax to the ground state.
However, this relaxation is almost always nonradiative. The fluorescence decay rate (rate of spontaneous
emission) of an excited state is related to the strength of the absorption that forms the excited state (see below).
- 104 -
If an absorption transition is very weak, such as the S
0
to S
1
transition in carotenoids, then the intrinsic
fluorescence decay rate constant will be very small, and fluorescence will make a negligible contribution to the
excited state decay. Internal conversion from S
1
to S
0
is typically rather efficient, so the S
1
state has a lifetime
in the ps-range.
Fig 5.8 Structures of several carotenoids and carotenoid precursors important in
photosynthetic systems
- 105 -
The energy of the S
1
state of the carotenoid is very difficult to measure directly, because of the forbidden
nature of the S
0
to S
1
transition. One method that can be used to determine this energy is detecting the (very
weak) fluorescence from S
1
. A more recent approach is two-photon spectroscopy, in which two photons from
a pulsed femtosecond are absorbed simultaneously, with the sum of their energies equal to the transition
energy. The S
0
to S
1
transition is allowed for two-photon absorption.
With both S
2
and S
1
having exceptionally short lifetimes, it is perhaps surprising that carotenoids are able to
carry out energy transfer to chlorophylls before they decay to S
0
, releasing their energy as heat. Yet in many
cases carotenoids are very efficient light-harvesting pigments, because the energy transfer process is even
(sometimes much) faster than the deactivation rate (see below).
Fig 5.9 Energy level diagram typical for carotenoids. The allowed
transition in normal light is to the S
2
state. Transitions to the S
1
state are forbidden because of symmetry. Excitation of S
2
is
followed by ultrafast (about 100fs) internal conversion to S
1
- 106 -
6. BioMolecules Viewed with Electronic Spectroscopy
In this chapter I will discuss the absorption and emission properties of biological molecules and
in particular those involved in photosynthesis: chlorophyll, bacteriochlorophyll, carotenoids. Due
to the fact that these molecules occur in so-called pigment protein complexes both their
interaction with the protein environment and the interaction with their neighbors gives rise to a
number of unique phenomena that play a crucial role in the process of photosynthesis. However,
photosynthetic pigments are not the only biomolecules that interact with light, another well-
known biological light-driven process is vision, carried out by the protein rhodopsin and the
pigment retinal, DNA-bases absorb in the ultraviolet region of the spectrum and electronic
spectroscopy is an easy tool to characterize the structure and dynamics of DNA. Finally
fluorescent markers can be attached to almost any protein, allowing them to be visualized by
microscopy and studied by single molecule techniques. The best known fluorescent marker that
can be attached by molecular genetics techniques to a selected protein is the green fluorescent
protein (GFP)
6.1 Absorption
The absorption of light by molecules gives them their color. The light that is not absorbed by the
substance can cause a sensation by being absorbed by the pigments in our eyes. So when we see
the characteristic color of a plant, it is due to the green light that was not absorbed. Light
absorption is at the heart of photosynthesis, so we will explore this phenomenon in a little more
depth. However, the absorption (and emission) properties of biomolecular systems are generally
very informative and can provide a wealth of information about the properties of biomolecules,
even in very complex environments (like in a living cell).
When light impinges on a collection of molecules, a number of processes may occur. First of all,
due to the interaction between the electro-magnetic wave and the matter, energy may be
absorbed or released. We will derive the equations governing the process, which will lead to the
well-known Einstein coefficients for absorption, stimulated- and spontaneous emission. Then we
will introduce the concept of optical density of a medium in connection with Lambert-Beers
law. Molecules have vibrations. When an electronic transition becomes excited, at the same time
a vibrational transition may be excited. This gives rise to vibronic progressions in molecular
absorption spectra. Finally, proteins are basically disordered. Although proteins can be
crystallized, and structures of proteins can be obtained, even up to high resolution, it is generally
accepted that the ground state of a protein is highly degenerate and that the protein may occur in
a large number of substates. In each of these substates the absorption properties of a protein-
bound pigment may be different and as a consequence the absorption spectrum of a pigment-
protein is highly inhomogeneous. This phenomenon is at the basis of techniques like spectral
holeburning, single molecule spectroscopy and photon echoes. At the end of this chapter we will
discuss some examples.
- 107 -
6.1.1 The transition dipole moment.
Consider a molecular (or atomic) system with discrete energy levels
k
E and wavefunctions
0
k
,
which are the unperturbed solutions of the time-independent Schrdinger equation of the
molecular system:
0 0 0
0 k k k
E H =
(6.1)
with
0
H the Hamiltonian that describes the molecular system. By switching on light with
frequency e we introduce a perturbation ( ) t H
1
, which starts to mix the eigenfunctions
0
k
. After
some time t the system will be found in a state ( ) t , which is now a solution of the time-
dependent Schrdinger equation
( ) ( ) ( ) t t H H
t
i
1 0
+ =
c
c
(6.2)
For a complete set of eigenfunctions
0
k
the time-dependent wavefunction ( ) t can be
expressed as
( ) ( )
t iE
k
k
k
k
e t c t
=
0
(6.3)
Insertion of this expression into Eq.6.2 yields a set of equations for the time derivatives of the
coefficients ( ) t c
k
given by
( )
( )
0
1
0
n k
t i
n
n
k
H e t c
i
dt
t dc
kn
e
(6.4)
with ( )
n k kn
E E = e . Taking
0
1
as the initial state, at t=0 we have ( ) 1 0
1
= c and
( ) ( ) 1 0 0 = = k c
k
. This allows us to integrate eq.6.4 for a time sufficiently short that
( ) ( ) 1 0 = ~ k t c
k
and ( ) 1
1
~ t c , which yields
( ) ' 1
1
0
'
1
dt H k e
i
t c
t
t i
k
k
}
=
e
(6.5)
The probability of finding our molecular system in the excited state k, thereby having absorbed a
quantum
1 k
e from the incident light beam is given by ( )
2
t c
k
. Eq.6.5 shows that ( )
2
t c
k
is
directly related to the quantity
2
1
1 H k . To obtain a useful expression for the transition
probability we have to find the form of the perturbation ( ) t H
1
.
An electromagnetic light wave is described by in-phase oscillating, perpendicular electric ( ) t E
and magnetic ( ) t B
fields that both obey a scalar wave equation provided that the velocity of the
electromagnetic wave is ( )
2 1
= c u . For a linearly polarized wave the electric field is given
by: ( ) t E t E e cos
0
= , where we have assumed that our molecule is in the origin of the coordinate
system and that its dimensions are much smaller than the wavelength . The response of the
- 108 -
molecule will be dominated by the interaction of the electric field with the electronic charge
distribution. For electrically neutral molecules the leading term in this interaction is the electric
dipole and thus
( ) ( ) t E t H
- =
1
(6.6)
where the electric dipole operator
i
i
i
r q
= in which the sum is taken over all the (partial)
electronic charges
i
q at positions
i
r
.
Substitution of eq. 6.6 into eq. 6.5, writing ' cos t e as the sum of two exponentials, performing
the integration and calculating the probability ( ) ( )
2
t c t P
k k
= that the system is in the state k at
time t yields
( )
( )
( )
2
2 2
2
0
2
2
2
1
sin cos
1
1
e e
e e u
=
kl
kl
k
t E
k t P
(6.7)
where we have assumed that the frequency of the light is close to the resonance frequency of the
transition k 1 , and with u the angle between the vectors
and
0
E
. In eq.6.7 we see that the
quantity
2
2
1
1
k
k
= determines the probability ( ) t P
k
: the larger
2
1 k
, the larger ( ) t P
k
and the stronger is the transition k 1 . The quantity
1 k
is called the transition dipole moment.
Note that the rate of light absorption due to the transition k 1 also depends on the orientation
of
1 k
relative to
0
E
via the term u
2
cos . For isotropic ensembles of molecules we will have to
average over all possible orientations of
1 k
relative to
0
E
. For an ordered system eq.6.7 leads to
polarization effects.
The probability ( ) t P
k
as expressed in eq.6.7 shows a marked frequency dependence, which in
addition depends on time. Intuitively we would like to associate the absorption of light energy to
a rate (i.e. a number of events per second) and this requires that the probability ( ) t P
k
is a linear
function of the time t. A closer inspection of eq.6.7 shows that as time proceeds the function
( ) t P
k
becomes sharply peaked around
kl
e e = . Furthermore, if we realize that in normal
spectroscopic experiments the incident light is far from monochromatic, it is more appropriate to
integrate eq.6.7 over a band of frequencies.
- 109 -
This leads directly to the rate of population of the level k:
( ) ( ) e e
c
t
W B W
dt
dP
k k
k
1
2
1
2
0
3
= =
(6.8)
where ( ) e W represents the (time-averaged) energy density of the incident electromagnetic field
at frequency e . Note that we have averaged eq.6.7 over all orientations of
1 k
relative to
0
E
,
which in 3-D space yields a factor 3 1 as the average over u
2
cos . The quantity
2
0
2
1
3
c t
ki k
B = is known as the Einstein coefficient for absorption and is directly related to
the extinction coefficient measured in a conventional absorption experiment.
Fig.6.1 Time and frequency dependence of P
k
as given by eq.6.7
- 110 -
When a beam of light impinges on a sample of (bio)molecules, part of
the light may be absorbed, the precise amount depending on the
wavelength. For instance, chlorophyll, the pigment of plants displays
two major transitions around 670 nm (red) and 430 nm (blue). In
addition several weaker transitions are visible.
( ) ( ) ( ) v u f E E dt dN
if if i
2 2 2
2
cos =
For a single molecule:
The quantity that determines the probability of a
transition to take place is the transition dipole
moment:
if
if
u
Where is a lineshape function ( ) v f
Absorption
6.1.2 The Einstein Coefficients
In this section we derive a simple relation between the rates of absorption, stimulated emission
and spontaneous emission in an atomic or molecular system, schematically depicted in 6.3. For
simplicity we assume that two energy levels exist,
1
E and
2
E , populated by N
1
and N
2
molecules, respectively. Three possible radiative processes which connect levels 1 and 2 are
indicated. Absorption and stimulated emission only occur with the light on and their rates are
given by ( ) e W B
12
and ( ) e W B
21
, respectively, in which
12
B is the Einstein coefficient for
absorption (eq.6.8) and
21
B is the Einstein coefficient for stimulated emission. We define
21
A as
the Einstein coefficient for spontaneous emission from level 2 to level 1 which may also occur in
the dark.
Fig 6.2 Summary of the process of absorption
- 111 -
For the rate of population of level 1 we have
2 21 2 21 1 12
1
N A WN B WN B
dt
dN
+ + =
(6.9)
Assuming a steady-state, i.e. 0
1
=
dt
dN
, it directly follows that
( )
( )
21 2 1 12
21
B N N B
A
W
= e
(6.10)
If we now take the situation that there is no external radiation field and that the system is in
thermal equilibrium at a temperature T, then the ratio
2 1
N N follows from the Boltzmann
distribution between states of different energies
kT
e
N
N
e
=
2
1
(6.11)
Under these conditions the energy density frequency distribution is given by Plancks radiation
law (averaged over polarizations).
( )
1
1
3 2
3
=
kT
e c
W
e
t
e
e
(6.12)
Since 6.10 should be equal to 6.12 it follows that
3 2
3
21
21
21 12
c B
A
B B
t
e
=
=
(6.13)
Using 6.8 we find
Fig.6.3 The three basic radiative processes. Indicated are the Einstein coefficients and the
transition rates for two levels with energies E
1
and E
2
and occupation N
1
and N
2
,
respectively.
- 112 -
2
12
0
3
3
21
3
c t
e
c
A =
(6.14)
The importance of this result is that the same quantity
2
12
determines the rates of all radiative
processes and consequently common selection rules apply to them. Moreover,
2
12
can be
obtained from a simple measurement of the absorption properties of a molecular system.
6.1.3 Lambert-Beers Law
The microscopic quantities
21 21 12 21
, , , A B B are connected to the macroscopic phenomenon of
light absorption by a colored sample via Lambert-Beers law. When a beam of light passes
through a sample of material, the light beam is usually absorbed, but sometimes amplified (in a
laser). Absorption dominates when most of the molecules are in their ground state ( )
2 1
N N >>
and when the intensity of the incident light beam is weak. In that case the rate equation (eq.6.9)
for transitions between states 1 and 2 is given by
( )
2 21 1 12
1
N A N W B
dt
dN
+ = e
(6.15)
Here ( )N W B e
12
is the rate at which light energy is removed from the incident light beam. Let the
light beam be propagated along the z-axis. Since the beam is attenuated its energy W will be a
function of z. Consider now a small slice of thickness dz and surface a as illustrated in fig 6.4.
The amount of energy in this slice in the interval e and e e d + is given by adz Wde . It then
follows from energy conservation that the rate of decrease of the beam energy must be equal to
the rate at which the energy is removed from the light beam by absorption. We find
( ) ( ) e e e e e d F
V
adz
W B N adz d
t
W
12 1
=
c
c
(6.16)
in which V adz represents the fraction of molecules in the selected slice adz , where V is the
volume of the sample. Moreover, we have assumed that not all the molecules absorb precisely at
Fig.6.4 Passage of a light beam through a thin slice of sample perpendicular
to the direction of the beam.
- 113 -
frequency
21
e , but that some statistical spread in transition frequencies exists at which the
molecules can absorb light (see below section 6.1.8: Linewidths). To account for this we
introduced ( ) e e d F , the fraction of molecules that absorb in the frequency interval e and
e e d + with
( ) 1 =
}
e e d F
(6.17)
Rearranging 6.15 gives
( ) ( )
V
W B F N
t
W e
e e
12 1
=
c
c
(6.18)
To obtain Lambert-Beers law we rewrite eq. 6.18 in terms of the change in intensity I in Wm
-2
of the light beam upon passage through the sample slice adz . Inspection of Fig.6.4 shows that
the difference between the amount of energy entering and leaving the sample slice per unit of
time precisely equals the rate of decrease of the beam energy. Thus we obtain
z I t W c c = c c . Moreover, cn I W = , with c the speed of light and n the index of refraction
of the medium. Substitution into 6.18 yields the required expression
( )
I
Vnc
F N B
z
I e e
1 12
=
c
c
(6.19)
The solution of eq.6.19 is straightforward and gives Lambert-Beers law, sometimes called
Beers law
( ) ( )
Kz
e I z I
= 0
(6.20)
with the absorption coefficient K given by
( )
Vcn
F B N
K
e e
12 1
=
(6.21)
Since
12
B was calculated from eq.6.8, in which integration over an absorption band was
performed, it follows that for any particular transition
12
B is independent of the frequency e .
From eqs.6.17 and 6.21 we find
e
e
d
K
N
Vcn
B
band
}
=
1
12
(6.22)
Thus, from a simple integration of the measurable(!!) absorption coefficient K over the
absorption line, we obtain the Einstein coefficient for absorption (and consequently the two other
Einstein coefficients). Moreover, since we have related
12
B to
2
12
the quantity
2
12
is
calculated/measured directly.
It is common practice to use instead of eq.6.20, the following expression for the dependence of I
on passage through a material with pathlength l:
( ) ( )
OD
I l I
= 10 0
(6.23)
where the optical density OD is defined as
lC OD c =
(6.24)
- 114 -
Here c is the molar extinction coefficient (usually expressed in dm
3
mol
-1
cm
-1
), l the pathlength
(usually in cm) and C the concentration of the sample (usually expressed in moldm
-3
). For
example, in a leaf chlorophyll a has in its absorption maximum at 680 nm an extinction
coefficient of 10
5
dm
3
mol
-1
cm
-1
. The chlorophyll concentration in a leaf is about 10
-3
mol dm
-3
and the pathlength is about 0.02 cm. Thus, the OD of a single leaf at 680nm is
2 10 2 10 10
2 3 5
= =
OD
. Thus, the reduction in intensity of 680 nm light upon passage
through a leaf is about a factor of 100. As a consequence no red light is detected below the outer
array of leaves of a tree.
It is not difficult to relate eqs. 6.23 and 6.24 to the expressions for K and B
12
and we finally find
for
2
12
( )
e
e
e c
d
band
}
=
61
2
12
10 01 . 1
(6.25)
where
2
12
is obtained in (Cm)
2
and all the other constants have been replaced by their
numerical values with the index of refraction 1 = n .
Returning to eqs.6.23 and 6.24 we can relate the extinction coefficient c to the absorption cross-
section of a single molecule o , given by P r
2
t o = , with r the radius of the molecule and P the
probability that a photon is absorbed upon passage through the surface
2
r t . Then
303 . 2
10
303 . 2
10
3 2 3
= =
A A
PN r N t o
c
(6.26)
in which N
A
is Avogadros number.
Light, Spectrum and Absorption
i
f
I
0
(v)
I(v)
( )
( )
( )
( ) ( )lC OD
I
I
OD
v c v
v
v
v
=
|
.
|
\
|
=
0
log
l
C =concentration
Beer-Lamberts law:
I
0
(v)
Fig.6.5 Summarizing Lambert-Beers law.
- 115 -
6.1.4 Franck-Condon factors
Molecules (and biomolecules) contain nuclei that vibrate relative to each other. Often these
nuclear motions are described by harmonic potential energy surfaces. When a molecule is
excited by light, the change in electronic state may be accompanied by a change in the
vibrational state of the molecule. This fact gives rise to a specific vibrational structure in the
electronic spectra of molecules. To understand these effects we must consider the nature of
nuclear motions during an electronic transition and this can be done by applying the Franck-
Condon principle, which states: Because the nuclei are so much more massive than the electrons,
an electronic transition takes place very much faster than the nuclei can respond. The quantum-
mechanical version of the Franck-Condon principle provides the correct picture. Before
absorption the molecule is in the lowest vibrational of its lowest electronic state (see fig.6.6), the
most probable location of the nuclei is at their equilibrium separation
e
R . The electronic
transition is most likely to take place when the nuclei have this separation. When the transition
occurs, the molecule is excited to the state represented by the upper curve. According to the
Franck-Condon principle the nuclear configuration remains unchanged during the electronic
transition, so we may imagine the transition as a vertical line in fig.6.6. The vertical line is the
origin of the expression vertical transition, which is used to denote an electronic transition that
occurs without a change of nuclear geometry.
- 116 -
The vertical transition cuts through several vibrational levels of the electronic excited state. The
level marked * is the one in which the nuclei are most probably at the same initial separation
e
R
(because the corresponding vibrational wavefunction has maximum amplitude there), so this
vibrational state is the most probable state for the termination of the transition. However, it is not
the only accessible vibrational state at the separation
e
R , because several nearby states have an
appreciable probability of the nuclei being at the separation
e
R . Therefore transitions may occur
(depending on the frequency of the incident light) to all vibrational states in this region, but most
intensely to the state with a vibrational wavefunction that peaks most strongly near
e
R . The
vibrational structure of the spectrum depends on the relative horizontal position of the two
potential energy curves, and a long vibrational progression, meaning a lot of vibrational
structure in the absorption spectrum, is observed if the upper potential energy curve is
appreciably displaced from the lower (see fig 6.7).The upper curve is usually displaced to greater
equilibrium bond lengths because electronic excited states usually have more antibonding
character than electronic ground states.
Fig 6.6 In the quantum mechanical version of the
Franck-Condon principle, the molecule undergoes a
transition to the upper vibrational state that most
closely resembles the vibrational wavefunction of
the lower electronic state. The two wavefunctions
shown here have the greatest overlap integral of all
the vibrational states of the upper electronic state
and hence are most similar
- 117 -
The quantitative form of the Franck-Condon factor is derived from the expression for the
transition dipole moment: i f
fi
= . The dipole moment operator is a sum over all the
:
+ =
k
k k
i
i
R Z e r e
(6.27)
where the vectors are the distances from the center of charge of the molecule.
Fig 6.7 Potential energy diagrams and spectra for absorption and fluorescence electronic
transitions in organic molecules. (a) The initial state for absorption is usually the ground
vibrational state of the ground electronic state, while the final state is usually an excited
vibrational state of the excited electronic state. (b) The initial state for fluorescence is the
ground vibrational state of the first excited state, while the final state is an excited
vibrational state of the ground electronic state. (c) Absorption and emission spectra that
result from the transitions shown in (a) and (b).
- 118 -
The overall wavefunction of a molecule (in the BO-approximation) consists of an electronic part,
e , and a vibrational part, v . Therefore, within the BO-approximation, the transition dipole
moment can be written as follows:
i k f
k
i f k i f i i
i
f
i i
i k
k k i f f fi
v R v e e Z e v v e r e e
v e R Z e r e v e
+
=
)
`
+ =
(6.28)
The second term on the second row of eq.6.28 is zero, because 0 =
i f
e e for two different
electronic states (they are orthogonal). Therefore,
( )
i f e e i f i i
i
f fi
v v S v v e r e e
i f
,
,
= =
(6.29)
where
i i
i
f e e
e r e e
i f
=
,
and ( )
i f i f
v v v v S = ,
The matrix element
i f
e e ,
is the transition dipole moment arising from the redistribution of
electron density upon the transition from i to f. The factor ( )
i f
v v S , is the overlap integral
between the vibrational state
i
v in the initial state of the molecule and the vibrational state
f
v in the final electronic state of the molecule.
We saw earlier that the transition intensity is proportional to the square of the magnitude of the
transition dipole moment, so the intensity of a vibronic absorption line is proportional to
( )
2
,
i f
v v S , which is known as the Franck-Condon factor for the transition. It follows that, the
greater the overlap of the vibrational wavefunction in the upper electronic state with the
vibrational wavefunction in the lower electronic state, the greater the absorption intensity of that
particular simultaneous electronic and vibrational transition. This conclusion is the basis of the
illustration in fig.6.6, where we see that the vibrational wavefunction of the ground state has the
greatest overlap with the with the vibrational states that have peaks at similar bond lengths in the
upper electronic state.
Exercise: Consider the transition from one electronic state to another, their equilibrium bond
lengths being
e
R and
'
e
R and their force constants equal. Calculate the Franck-Condon factor
for the 0-0 transition and show that this transition is most intense when the equilibrium bond
lengths are equal.
- 119 -
6.1.5 The electronic spectra of polyatomic molecules
Absorption of a photon can often be traced to the excitation of specific types of electrons or to
electrons that belong to a small group of atoms in a polyatomic molecule. For example when a
carbonyl group (>C=O) is present (like in the peptide bond of proteins) an absorption at about
290 nm is normally observed, although its precise location depends on the nature of the rest of
the molecule (o-helix vs. |-sheet vs. random coil). Groups with characteristic optical absorption
are called chromophores (from the Greek word for color bringer) and their presence often
accounts for the colors of substances. Other examples of chromophores are a base in DNA or a
chlorophyll molecule in a chlorophyll-protein complex.
t->t- and n->t* transitions
Absorption of light by a C=C double bond excites a t electron into an antibonding t* orbital
(fig.6.8).
The chromophore activity is therefore due to a t->t- transition. Its energy is about 7 eV for a
single conjugated double bond (like in ethene), which corresponds to an absorption at 180 nm (in
the ultraviolet). When the double bond is part of a conjugated chain (like in a carotenoid), the
Fig 6.8. The C=C double bond acts
as a chromophore. One of its
important electronic transitions that
can be driven by light is the t>t*
transition illustrated here, in which
an electron is promoted from a t-
orbital to the corresponding
antibonding orbital by the absorption
of a photon.
- 120 -
energies of the molecular orbitals lie closer together and the t->t- transition moves to longer
wavelength; it may even lie in the visible region if the conjugated system is long enough. An
important example of a t->t- transition is provided by the pigment retinal that plays the
essential role in the photochemical mechanism of vision (see 6.1.7).
In a simple molecule like formaldehyde (H
2
CO) we can distinguish another, weaker transition to
the lower energy than the t->t- transition. It originates from the lone pair of electrons on the O-
atom. The concept of a lone pair refers to pairs of electrons in an orbital that is largely confined
to one atom and not appreciably involved in bond formation. One of these electrons may be
excited into an empty t* orbital of the carbonyl group, which gives rise to a n>t- transition.
Typical absorption energies are about 4 eV (290 nm). Because n>t- transitions in carbonyls are
symmetry-forbidden, the corresponding absorptions are weak. They become allowed due to
deformations of the molecule
Exercise 6.1
Give the electronic structure and energy-level diagram for H
2
CO. Show that in H
2
CO the t>t-
transition is polarized along the axis connecting the C and O atom. Show that the n>t-
transition in formaldehyde is symmetry-forbidden.
Exercise 6.2
Show that the electronic transitions of aromatic molecules are in the plane of the molecule
Exercise 6.3
Show (using the Free Electron Molecular Orbital model) how the lowest energy transition for
long conjugated chains shifts to the red for longer conjugated chains.
- 121 -
6.1.6 Linear Dichroism
Since the transition dipole moment has a well defined and known orientation in the frame of the
molecule, one can use polarized light to find out how (bio)molecules and even complex
molecular aggregates are organized in space. Suppose you have a DNA molecule and suppose
that it would be possible to measure the absorption of this DNA molecule with a light beam that
is polarized parallel to the helix axis and with a light beam that is polarized perpendicular to the
helix axis. In case the DNA bases are truly perpendicular to the helix axis, the first beam would
be transmitted for 100%, while the second beam would be partially absorbed. This is due to the
fact that the transition dipoles of all the DNA bases are in the plane of the base, and consequently
perpendicular to the helix axis.
In a sample that contains a random distribution of DNA molecules this effect would be lost,
because DNA molecules occur with all possible orientations. However, if one would be able to
orient the DNA molecules in some known manner, then, depending on how well one could orient
the DNA molecules, a difference in the absorption between light polarized parallel and light
polarized perpendicular to some orientation axis would be measured (see fig. 6.9 and 6.10). This
phenomenon is called linear dichroism and the amount of linear dichroism is a direct measure for
the three dimensional structure of a biomolecule containing light absorbing groups.
Similarly in a pigment protein one can measure the angle between the transition dipole moment
of the chromophores and the orientation (symmetry) axis.
Linear dichroism (1)
E
par
E
perp
Perfectly oriented DNA
Fig. 6.9 Illustration of a polarized absorption measurement on oriented DNA
- 122 -
DNA and long biomolecules display large inducible dipoles moments when exposed to an
externally applied electrical field. As a consequence they will align with their long axis along the
electric field lines. One disadvantage of this kind of orientation is that it can only be applied
under low salt conditions to avoid high currents. Another way of aligning biomolecules is by
inserting them into a deformable polymer gel (gelatin, polyacrylamide) and compressing the gel
in two directions while allowing it to expand into the third direction. As a consequence (large)
biomolecules will be oriented. Long DNA and protein molecules will be oriented with their long
axis parallel to the axis along which the gel expands, disk-shaped proteins, such as
photosynthetic membrane proteins will orient with the normal to the disk perpendicular to the
expansion axis.
Linear Dichroism (2)
Fig 6.10 Illustration of the linear dichroism experiment on an
oriented ensemble of (bio-)molecules.
- 123 -
The amount of absorption for light incident along the y-axis and polarized along the x-axis is
given by:
x x
k A u
2 2
12
cos = (6.30)
where k is some proportionality constant and ..... reflects averaging over all chromophores
within one biomolecule and averaging over all possible orientations. In case there is no net order
in the system:
3
1
cos cos cos
2 2 2
= = =
z y x
u u u (6.310
Then:
( )
2
12
3 1 k A A A A
iso z y x
= = = = (6.320
With A
iso
the absorption of an isotropic sample measured with unpolarized light. With the light
incident along the y-axis we can define the amount of dichroism as:
x z
A A A = A (6.33)
And the reduced dichroism as:
( )
x z iso x z r
A A A LD u u
2 2
cos cos 3 = = (6.34)
Linear Dichroism (3) How to orient
biomolecules?
Fig 6.11 How to orient biomolecules? By putting them into a gel and compressing the
gel along the x,y directions and allowing it to expand in the z-direction rod-like
biomolecules will be oriented along the long axis of the gel.
- 124 -
For a disordered sample we find of course: ( ) 0
3
1
3
1
= =
r
LD , while for a sample with all the
transition dipoles perfectly oriented along the z-axis we find: ( ) 1 0 1 = =
r
LD . Thus LDr is the
quantity of interest and contains the information about the molecule that is being studied. In
principle the angle between the transition dipole and the symmetry-axis of the particle can be
obtained. The unknown parameter is the degree of orientation, for which some model must be
developed.
Linear Dichroism (4): Perfectly ordered rods
u
u
u
We have a sample of perfectly
ordered rod-shaped molecules.
The molecules contain one
coloured group, a
chromophore, which has its
transition dipole
12
at an
angle u with the long axis of
the molecule.
y
x
z
12
12
12
u u
u
t
2 2
12
2
2
0
2 2
12
2 2
12
sin
2
1
cos sin
cos
= =
=
}
d k A
k A
x
z
Then we have:
( )
( ) 1 cos 3
2
1
sin cos
sin
2
1
cos
3
2
2 2
2 2
=
+
= u
u u
u u
iso
x z
r
A
A A
LD
Thus, for the reduced linear
dichroism:
Note that there exists an angle u
M
for which LD
r
=0, in spite of the fact that the sample
is perfectly ordered!!
3 1 cos
2
=
M
u
or
o
M
7 . 54 = u : The magic angle
Rod-like biomolecules can easily be oriented in an electrical field (assuming they have an
inducible dipole moment, which is of course almost always the case), in a stretched film and in a
gel. They will orient with their long axis along the orientation direction (the electric field lines or
the stretching direction of the film/gel). Disk-like particles like photosynthetic membrane
proteins will orient with the normal to the plane of the disk perpendicular to stretching direction.
Fig 6.12 The Linear Dichroism of perfectly oriented rod-like biomolecules (long proteins, DNA,
..)
- 125 -
Linear Dichroism (5) The o-helix
Elements of secundary structure: the o-
helix
t
//
t
Fig 6.13. Linear dichroism of oriented poly-L-glutamic acid. This o-helical synthetic
polypeptide is oriented by putting it into a film and stretching the film. Bands corresponding
to perpendicular n->t* and t>t- transitions and parallel t>t* transitions are indicated.
Note how the t>t* transition around 200 nm is split into a perpendicular part to the blue
of the original transition, and a parallel part to the red of the original transition. This
originates from the coupling between t>t* transitions on adjacent peptide bonds in the o-
helical structure. (see the section on excitons)
- 126 -
Linear Dichroism(6) The degree of orientation
In general one can not obtain a perfect orientation of the molecule under study. For
instance in a pressed gel one typically reaches about 10-20%, DNA in a flow cell
>50%, membranes spread out on a surface 30% etc. In that case it is useful to define a
function u which represents the degree of orientation and investigate by independent
means how large u is. In general we have:
In which <....> indicates the averaging of the angles that occur in one particle, and u
is a function that rangs between 0 (no orientation) and 1 (perfect orientation).
An example: for a gel which is compressed in both the x and y direction with a factor
and expands in the z-direction with a factor n one can derive that:
( ) u = 1 cos 3
2
1
2
u
r
LD
u
n
( ) ( ) | |
( ) ( ) | | ( ) ( ) ( ) ( ) 1 1 arctan 1 1
1 3
2
1
3 3 3 3
=
= u
n n n n n T
n T n
For instance, gels can be compressed so much that their length in the z-axis increases by
about a factor of 2.
Fig 6.14. How the linear dichroism of an oriented ensemble of biomolecules depends on
the degree of orientation
- 127 -
6.1.7. The photochemistry of vision
The eye is an exquisite photochemical organ that acts as a transducer, converting radiant energy
into electrical signals that travel along neurons to the brain. Here we concentrate on the events
taking place in the human eye, but similar processes occur in all animals. Indeed, a single type of
protein, rhodopsin, is the primary receptor for light throughout the animal kingdom, which
indicates that vision emerged very early in evolutionary history, no doubt because of its
enormous value for survival.
Photons enter the eye through the cornea, pass through the ocular fluid that fills the eye, and
Fig.6.15.Artists view of the structure of the visual protein rhodopsin. Rhodopsin is a
membrane protein, consisting of 7 transmembrane helices (purple) connected by loops. The
protein provides a binding site for the chromophore retinal (in light blue), which absorbs
the light and triggers the conformational change that eventually leads to a signal to the
brain. The retinal molecule is covalently attached to a lysine residue via a so-called Schiffs
base. Note the chain of water molecules (black) that connects the chromophore with the
membrane-water interface. Proton rearrangement in this chain of water molecules is an
essential part of transmitting the light-induced conformational change of the retinal to the
outside world. Rhodopsin is part of the family of G-type receptors, which are structurally
very similar to rhodopsin, but all the other members of the family have no chromophore.
They recognize compounds like hormones at the outside of the cell and transmit the signal
to the inside. In that sense, rhodopsin recognizes photons!!!
- 128 -
fall on the retina. The ocular fluid is principally water and passage of light through this medium
is largely responsible for the chromatic aberration of the eye, the blurring of the image as a result
of different frequencies being brought to slightly different focuses. Water, like any other
transparent medium, disperses light because of the fact that its index of refraction varies with the
frequency. This effect can be traced back to the connection between polarizability and refractive
index: the greater the polarizability, the greater the refractive index and the greater the bending
of a ray of light. Water is more polarizable in response to blue (high-frequency) light than to red
(low-frequency) light because each photon of blue light has more energy and can more readily
interact with the water molecules. The chromatic aberration is reduced to some extent by the
tinted region called the macular pigment that covers part of the retina. The pigments in that
region are the carotene-like xanthophylls, which remove some of the blue light and hence help to
sharpen the image. They also protect the photoreceptor molecules from too great a flux of
potentially dangerous high-energy photons. The xanthophylls have delocalized electrons that
spread along the chain of conjugated double bonds, and the t-to-t* transition lies in the visible.
Much of the same type of photoprotection is found in the pigment-proteins that occur in
photosynthesis, where carotene molecules help to protect the delicate (and much more reactive)
chlorophyll molecules.
About 57% of the photons that enter the eye reach the retina; the rest are scattered or absorbed by
the ocular fluid. Here the primary act of vision takes place, in which the chromophore of a
rhodopsin molecule (rhodopsin is a membrane protein) absorbs a photon in another t-to-t*
transition. A rhodopsin molecule consists of an opsin protein molecule to which is attached an
11-cis-retinal molecule. The later resembles half a carotene molecule, showing Natures
economy in its use of available materials (and available biosynthetic pathways!!!). The
attachment of the retinal to the opsin is by the formation of a Schiffs base, utilizing the CHO
group of the chromophore and the NH
2
group from a lysine side-chain. The free 11-cis-retinal
molecule absorbs in the UV, but the attachment to the opsin protein molecule shifts the
absorption into the visible region. The rhodopsin molecules are situated in the membranes of the
rods and cones that cover the retina. The opsin molecule is anchored into the cell membrane by
two hydrophobic groups and largely surrounds the retinal chromophore (see fig 6.15).
Immediately after the absorption of a photon, the 11-cis-retinal molecule undergoes
photoisomerization into all-trans-retinal (see fig 6.16). Photoisomerization takes about 200 fs and
about 67 retinal molecules isomerizes for every 100 photons that are absorbed (in other words:
the quantum yield is 67%). The process is able to occur because the t-to-t* excitation of an
electron loosens one of the t-bonds (the one indicated by the arrow in the diagram), its torsional
rigidity is lost, and now one part of the molecule swings round to its new position. At that point
the molecule returns to the ground state, but now trapped in its new conformation. The
straightened tail of all-trans-retinal results in the molecule taking up more space than the original
11-cis-retinal did, so the molecule exerts a force on the protein structure of the opsin protein that
surrounds it. Thus in about 0.25-0.5 ms after the initial absorption event, the rhodopsin molecule
is activated.
Now a sequence of (bio-)chemical events the biochemical cascade converts the energy
accumulated in the activated form of the rhodopsin molecule into a pulse of electric potential that
travels through the optical nerve into the optical cortex, where it is interpreted as a signal and
- 129 -
incorporated into the web of events that we call vision. At the same time the resting state of the
rhodopsin molecule is restored by a series of non-radiative chemical events powered by ATP.
The process involves the escape of all-trans-retinal as all-trans-retinol (in which CHO has been
reduced to CH
2
OH) from the opsin molecule by a process catalyzed by the enzyme rhodopsin
kinase and the attachment of another protein molecule, arrestin. The free all-trans-retinol
molecule now undergoes enzyme-catalyzed isomerization into 11-cis-retinol followed by
dehydrogenation to form 11-cis-retinal, which is then delivered back into an opsin molecule. At
this point, the cycle of excitation, photoisomerization, and regeneration is ready to start all over
again.
Fig. 6.16 Vision
- 130 -
6.1.8 Linewidths and lineshapes
Molecular spectral lines are not infinitely narrow, but display a certain width e A . A large
variety of phenomena and processes may contribute to the observed line widths and here we will
discuss a few, assuming a single type of molecule. In principle we distinguish homogeneous and
inhomogeneous broadening. In the first case all the molecules in an ensemble that contribute to
an absorption line suffer from the same broadening of the line. In inhomogeneous broadening
different molecules in the ensemble absorb at (slightly) different frequencies, which results in a
line broadening of the sample as a whole.
Homogeneous broadening
We first discuss lifetime broadening as a form of homogeneous broadening. If the excited state
produced by the absorption of electromagnetic radiation has a finite lifetime t due to the
possibility of spontaneous emission, internal conversion, excitation energy transfer,
photochemistry etc. the correct expression for the excited state wavefunction is:
( ) ( ) ( ) ( ) t 2 exp 0 , , t t iE r t r
f f f
=
(6.35)
We may express the time-dependent part of ( ) t r
f
,
as a linear combination of pure oscillating
functions of the type ( ) ( ) Eit exp by means of a Fourier transformation and obtain
( ) ( ) ( ) ( ) ( )dE t iE E L t t iE
f
=
}
exp 2 exp t (6.36)
in which ( ) E L is the distribution or line shape function. Eq.6.36 demonstrates that the finite
excited state lifetime t implies that we have an uncertainty in the excited state energy, reflected
by the distribution function ( ) E L , which can be easily calculated to have the form
( )
( ) ( )
2 2
2t
t
+
=
E E
E L
f
(6.37)
The line shape represented by eq.6.37 is called a Lorentzian. The function has a sharp peak
around
f
E E = but falls off slowly at higher and lower energies. The full width at half-maximum
(FWHM) of the Lorentzian is given by:
t = AE or t e 1 = A (6.38)
Note that eqs.6.38 are strongly reminiscent of Heisenbergs uncertainty relation and therefore
lifetime broadening is often referred to as uncertainty broadening.
For atoms/molecules where the excited state lifetime is only determined by spontaneous
emission the lifetime is called the natural or radiative lifetime,
R
t , given by
- 131 -
A
R
=
1
t (6.39)
Where A is the Einstein coefficient for the transition, representing the number of spontaneous
emission transitions per second. The resulting natural or radiative linewidth is the minimum
linewidth. For atoms in the gas phase and for complex molecules (like chlorophyll) at 0 ~ T ,
where other decay processes no longer contribute,
1
R
t may be the experimentally observed
linewidth. Note that the natural linewidth depends strongly on the emission frequency e through
the Einstein coefficient (eq. 6.34). Since they increase with
3
e , electronic transitions will have
significant natural linewidths, while vibrational (and for small molecules rotational) transitions
will be very narrow. Typically, for an allowed transition the natural lifetime may be of the order
of s
8 7
10 10
, corresponding to a natural linewidth t e v 2 A = A of Mhz 15 5 . 1 . In contrast, a
typical natural lifetime for a rotational transition is about
3
10 s, corresponding to a natural
linewidth of only . 10 5 . 1
4
Hz
-
Normally for atoms and molecules many processes may contribute to the shortening of the
lifetime t and hence increase the linewidth of an absorption band. Let us discuss
atoms/molecules in the gas phase, or molecules in a solvent, which may undergo many
collisions, either with other atoms/molecules of with solvent molecules. One usually assumes
that the phases of the excited state wavefunctions before and after the collision are totally
uncorrelated. This means that the wavefunction no longer has the form of a pure wave train, only
disturbed by the natural lifetime. If the lifetime is long compared to the timescale of the
collisions one may approximate the wave train by a pure wave with one frequency only. Because
of the collisions between atoms/molecules and their surroundings, the wave train is chopped into
portions of length
coll
t . The relative phases of these portions will be completely random.
Collisions in this way produce a dephasing of the wave function. One may show that this will
result in a Lorentzian line shape of the transition with the FWHM given by:
coll
coll
t
e
2
= A (6.40)
Where
1
coll
t is the rate at which the collisions occur.
Molecules in the condensed phase (in solution, or in a host matrix) will interact with their
surroundings and collisions will occur between the molecule and phonons in the medium, also
leading to dephasing of the excited state. The homogeneous line width
hom
v A
of a molecular electronic transition is often expressed as
*
2 1
hom
1
2
1
T T t t
v + = A (6.41)
Where
1
T is the observed excited state life time and
*
2
T is the pure dephasing time. Note that
1
T
is generally much smaller than the natural life time
R
t , since in molecules many processes may
contribute to the decay of the excited state. Spectral hole-burning, where homogeneous line
- 132 -
widths are measured as a function of temperature, is a technique to extract these two parameters
1
T and
*
2
T .
Inhomogeneous broadening
Inhomogeneous broadening of spectral lines occurs when different molecules or molecular
environments contribute to slightly different parts of the absorption spectrum. An example for
atoms and small molecules is the Doppler broadening, caused by the fact that in the gas phase the
atoms/molecules do not all have the same velocity, but their velocities are distributed around a
certain average value. In the case of emission the Doppler shift of the emission wavelength
depends on the velocity component in the direction of the detector. A simple calculation based
on the Maxwell distribution yields a Gaussian line shape function.
For molecules in a non-gaseous environment the interaction of the molecule with the host will
fine-tune their absorption frequencies. For a crystalline host this may give rise to a (limited) set
of relatively sharp, well-defined lines in low-temperature spectra. For molecules in a solution or
in a glassy environment (such as a protein) a broad distribution of sites may exist, each with their
own transition frequency, leading to the broad (
1
50
~ A cm v ) unstructured inhomogeneously
broadened absorption bands observed for many (bio)molecules amongst which are the natural
pigments, such as chlorophyll and |-carotene. Specific spectroscopic techniques such as spectral
holeburning, fluorescence line-narrowing and single molecule fluorescence spectroscopy (see
fig. 6.17) have to be applied to resolve the underlying homogeneous line widths and
inhomogeneous distribution function.
Single Molecule Spectroscopy(1)
- 133 -
Composite Line Shapes
When two independent processes contribute to the line shape, the resulting spectral line will be
given by a so-called convolution of both line shapes
( ) ( ) ( ) ' ' '
0 2 1
e e e e e e d L L L + =
}
(6.42)
where
0
e is the common central frequency of the two individual line shape functions.
Appendix Ch. 6 Collision broadening
The collisions between atoms in the gas phase (or molecules and the solvent) is an important
source of line broadening. Take a particular atom radiating at frequency
0
e . One can envisage a
wave train of electromagnetic radiation steadily emanating from the atom until the atom make a
collision. During the collision the energy levels of the radiating atom are shifted by the forces of
interaction between the two colliding atoms. Thus the radiated wave train is interrupted for the
duration of the collision. When the wave of frequency
0
e is resumed after the collision, its
characteristics are identical to those which it had prior to the collision, except that the phase of
the wave is unrelated to the phase before the collision.
If the collision is sufficiently brief, it is possible to ignore any radiation emitted during the
collision while the frequency is shifted away from
0
e . The collision broadening effect can then
be adequately described by a model in which each excited atom always radiates at frequency
0
e
but with random changes in phase of the radiated wave each time a collision occurs. The
apparent spread in emitted frequencies arises because the wave is chopped into finite sections
whose Fourier decompositions include other frequencies than
0
e .
Fig 6.17. Single molecule fluorescence excitation spectra of the bacterial light-harvesting
complex LH2 (bottom left). The ensemble spectrum (top right) of this complex displays broad
absorption peaks at 800 nm (due to the green bacteriochlorophyll molecules, which are only
weakly coupled to all the others) and at 850 nm (due to the blue bacteriochlorophylls, which
are strongly coupled). On the bottom right excitation spectra are shown for 5 single LH2s.
Note the sharp lines in the 800 nm region, which reflect single green bacteriochlorophyll
molecules in the LH2-protein environment. Note that the linewidths in the 850 nm region are
much broader, reflecting the lifetime broadening due to femtosecond relaxation processes
occurring between the different exciton states.
- 134 -
The wave train radiated by a single atom is illustrated schematically in the next
figure.
Which shows the variation of the electric field amplitude ( ) t E at a fixed observation point as a
function of time. The occurrence of a collision is represented by a vertical line accompanied by a
random change in the phase of the wave. According to the kinetic theory of gases, the probability
( ) t t d p that an atom has a period of free flight lasting a length of time between t and t t d + is
( ) ( ) ( ) t t t t t t d d p
0 0
exp 1 =
With the mean time of free flight
0
t given by:
2
1
2
0
4 1
|
|
.
|
\
|
=
M V
N d
|
t
t
Here d is the distance between the centers of the atoms during the collision, N is the number of
particles, V is the volume, M is the atomic mass and T k
B
= | .
The periods of free flight in the above figure are chosen in accordance with the probability
distribution. The variation of the phase is with time is shown below
- 135 -
Consider a period of free flight of an atom, which commences at time
0
t and lasts a time t . The
field amplitude in complex form can be written as:
( ) ( ) | e i t i E t E + =
0 0
exp
Where | is the phase of the wave for the particular period of free flight considered, but
0
E and
0
e are the same for any period. The field ( ) t E can be represented as a Fourier integral where the
amplitude at frequency e is given by
( ) ( )
( ) { }
( ) { }
( )
0
0
0 0
0
0 0
1 exp
exp
2
exp
2
1
0
0
e e
t e e
| e e
t
e | e
t
e
t
+
= + + =
}
+
i
i
i t i
E
dt t i i t i E E
t
t
.
This a cycle averaged intensity of the radiation emitted during the period has a frequency
distribution
( ) ( )
( ) { }
( )
2
0
0
2
2
0
2 2 sin
e e
t e e
t
e e
|
.
|
\
|
=
E
E I
At any instant in time the total intensity of radiation is made up of contributions from a large
number of excited atoms. The times of free flight of the different atoms are distributed according
- 136 -
to ( ) ( ) ( ) t t t t t t d d p
0 0
exp 1 = , and it is necessary to weigh each contribution ( ) e I with the
probability ( ) t p and integrate over t to find the total intensity:
( )
( ) { }
( )
( )
( ) ( )
2
0
2
0
0
0
2
0
0
2
0
1
2
1
exp
2 sin 1
t e e
t t t
e e
t e e
t
e
+
=
d I
total
The frequency distribution of a collision-broadened emission line thus has a Lorentzian shape
with width
0
2 t , that is twice the mean rate at which collisions occur.
- 137 -
Spec.1.9. Some Examples.
1. Chlorophyll
Below the absorption spectrum of chlorophyll is shown together with the polarization direction
of the Q
y
and Q
x
transitions
The absorption spectrum of chlorophyll
Chlorophyll a Chlorophyll a
670nm
630nm
Fig. 6,18. The absorption spectrum of chlorophyll a. There are two major transitions in
the molecular frame: the Q
y
transition at 670-680 in red and the perpendicular Q
x
transition in orange. The transitions in the blue have a mixed polarization. Also note the
vibrational progression on the Q
y
transition.
- 138 -
2.The bacterial light-harvesting complex LH2
In fig 6.20 we show the absorption spectrum of a bacterial light-harvesting complex, LH2, which
collects light and transfers electronic excitations to the bacterial photosynthetic reaction center,
where the electronic excitation energy is converted into a charge separation. The complex
contains bacteriochlorophylls and carotenoids as pigments. Both are very strongly absorbing
molecules with extinction coefficients between 100.000-150.000 dm
3
mol
-1
cm
-1
. Monomeric
bacteriochlorophyll in organic solvents absorbs around 770 nm. In the LH2 complex pigment-
pigment interactions have shifted the main band to 850 nm. Carotenoids absorb in the visible,
their color depends amongst others on the number of conjugated carbon atoms, and they are the
major light-harvesters, transferring the absorbed solar energy with >90% efficiency to
bacteriochlorophylls.
Fig 6.19. A sample of chlorophyll-
proteins is illuminated with blue
light (a blue laser beam passing
through the sample). The Soret
levels of the chlorophylls are
excited. Rapid (100 fs) internal
conversion to the Q
y
level occurs
and on a ns timescale the red
fluorescence is emitted.
- 139 -
Fig6.20. Top: Peripheral bacterial light-harvesting complex (LH2). 24 bacteriochlorophylls and
8 carotenoids are non-covalently bound to two concentric rings of membrane-spanning o-
helices. The top 16 bacteriochlorophylls form a strongly coupled ring in which the pigments
interact strongly giving rise to a red-shifted absorption band at 850 nm. The bottom ring consists
- 140 -
of 8 bacteriochlorophylls which are weakly coupled and which absorb around 800 nm. A
weaker transition of both rings is observed around 580-600 nm.
The 8 carotenoids span the membrane and absorb in the region 440-570 nm.
Bottom: the dashed curve is the absorption spectrum of LH2, with pronounced absorption bands
at 850 nm, 800 nm, 590 nm due to the bacteriochlorophylls and bands at 550, 515 and 480 nm
due to the carotenoids. The intense peaks around 350-400 nm also belong to the
bacteriochlorophylls. A solar photon absorbed by one of the carotenoids is very efficiently
transferred to either the green bacteriochlorophylls, or the blue bacteriochlorophylls.
Excitations in the green ring are subsequently transferred to the blue ring. In the
photosynthetic membrane, LH2 is coupled to LH1, another light-harvesting complex that
absorbs more to the red (880nm) and which feeds excitations into the photosynthetic reaction
center, where they are converted into a charge separation. The spectrum of LH1 is given by the
drawn curve.
3. The Bacterial Reaction Center
The Bacterial Reaction Center
Example 4. The eye-lens protein o-crystallin and DNA
Fig 6.21 The bacterial reaction center is a pigment-protein complex that is excited by energy
transfer from bacteriochlorophylls in the LH1 antenna complex or by direct absorption of a
photon. Following excitation an electron is transferred from one side of the photosynthetic
membrane to the other (6.21 right). Only the pigments in the A (or active) branch are involved in
this charge separation process. The left shows the characteristic absorption spectrum of the
bacterial reaction center. The band at about 880 nm labeled P originates from the special pair of
bacteriochlorophylls (blue) in the heart of the complex. The band at about 800 nm originates from
the two accessory bacteriochlorophylls (green) labeled B
A
and B
B
. The band at about 760 nm is
due to the two bacteriopheophytins H
A
and H
B
.
- 141 -
Fig. 6.22 shows the solar emission spectrum below 340 nm at the earths surface in combination
with the absorption spectrum of two important biomolecules: DNA, the carrier of the genetic
code and o-crystallin, the major protein of the mammalian eye lens.
Fig 6.22. Absorption spectra of DNA (dot-dashed) ) and o-crystallin (dashed) in the wavelength
region 240-340 nm. The solid line indicates the solar emission spectrum at the earths surface.
The absorption of light by these biological molecules is essentially zero in the region 320nm <
<400 nm, which is called near UV or UV-A, intense in the region 200-280 nm, which is called
the far-UV or UV-C and only overlaps with the solar spectrum in the wavelength region 290nm
< < 320nm, the mid-UV or UV-B.
The absorption of DNA is due to the aromatic DNA bases guanine, thymine, cytosine and
adenine and it peaks at about 260 nm, with a maximum extinction of
1 1 3 4
10
= cm mol dm c
(expressed per mole of base). The electronic transitions which contribute to the absorption in the
220-290nm wavelength region are predominantly
* t t
transitions oriented in the plane of
the DNA base. The absorption of proteins like o-crystallin is mainly due to the aromatic amino
acids tryptophan and tyrosine and it peaks at about 280 nm. For a protein with nTRP tryptophan
amino acids and nTYR tyrosine amino acids the extinction coefficient at 280 nm is given by
1 1 3
280
1100 5600
+ ~ cm mol dm n n
TYR TRP
c
For a typical biological cell the total absorption of solar UV due to proteins would be about 10%
of the absorption due to nucleic acids.
Often proteins carry cofactors which allow proteins to carry out their specific tasks. For instance,
in hemoglobin, the heme group, a porphyrin molecule, is attached to the protein in a heme-
protein complex, which is active in oxygen binding and release in blood. In those cases the
absorption extends into the near-UV, visible and sometimes near-infrared region of the spectrum.
- 142 -
In several cases the absorption of light in this wavelength range is crucial for the biological
function (photosynthesis, vision).
- 143 -
6.2 The Fates of the Electronic Excited States
A radiative process is a process in which a molecule discards its excitation energy as a photon. A
more common fate is nonradiative decay, in which the excess energy is transferred into vibration,
rotation, and translation of the same or of surrounding molecules. This thermal degradation
converts the electronic excitation energy completely into thermal motion of the environment
(that is, to heat). An excited molecule may also take part in a chemical reaction. For instance,
in photosynthesis, following the light absorption and energy transfer process, a special pigment
in the photosynthetic reaction center becomes electronically excited that then drives a very
efficient charge separation, by which electronic excitation energy is converted into the energy of
a charge separated state.
6.2.1 Fluorescence and Phosphorescence.
The J ablonski Diagram
In fluorescence, spontaneous emission of radiation occurs within a few nanoseconds after the
exciting laser pulse. In phosphorescence the spontaneous emission may persist for long times
(seconds, even hours). The difference suggests that fluorescence is a fast conversion of absorbed
radiation into re-emitted photons, while phosphorescence involves the storage of energy into a
new electronic state from which it slowly leaks.
Fluorescence
Fig. 6.23 shows the sequence of steps involved in fluorescence. The initial absorption takes the
molecule to an electronic excited state, and if the absorption spectrum were monitored, it could
look like the one shown in fig. 6.23 . The excited molecule is subjected to collisions with the
surrounding environment (for a pigment in a solvent this would be solvent molecules, for a
pigment in a protein this would be the protein environment), and as it gives up energy
nonradiatively it steps down the ladder of vibrational states to the lowest vibrational level of the
electronically excited molecular state Typically this is a fast process, 100 fs-1 ps. The
surrounding molecules, however, might now be unable to accept the larger energy difference
needed to lower the molecule to the ground electronic state. It might therefore survive long
enough to undergo spontaneous emission (with a rate constant given by the Einstein coefficient
for spontaneous emission A
21
) and emit the remaining excess energy as radiation. The downward
electronic transition is vertical (in accord with the Franck-Condon principle) and the
fluorescence spectrum has a vibrational structure characteristic of the lower electronic state.
- 144 -
Radiationless decay and fluorescence
10
-12
s
10
-9
s
Provided that they can be seen, the 0-0 absorption and fluorescence transitions can be expected
to coincide. The absorption spectrum arises from 0-0, 1-0, 2-0 transitions and occur at
progressively higher frequencies and with intensities governed by the Franck-Condon principle.
The fluorescence spectrum arises from 0-0, 0-1, 0-2 downward transitions, and hence occur
with decreasing frequencies. The 0-0 absorption and fluorescence peaks are not always exactly
coincident because the solvent may interact differently with the molecule in the ground and
excited states (for instance the H-bonding pattern may be different). Because the solvent
molecules do not have the time to rearrange during the transition, the absorption occurs in an
environment characteristic of the solvated ground state; however, the fluorescence occurs from
in an environment characteristic of the solvated excited state (see fig 6.24).
Fig 6.23 Left: The sequence of steps leading to fluorescence. After the initial absorption, the
upper vibrational states undergo radiationless decay by giving up energy to the
surroundings. A radiative transition then occurs from the vibrational ground state of the
upper electronic state. Right: Absorption spectrum (a) shows a vibrational structure
characteristic of the upper state. A fluorescence spectrum (b) shows a structure
characteristic of the lower state; it is also displaced to lower frequencies (but the 0-0
transitions coincide) and resembles a mirror image of the absorption
- 145 -
Stokesshift:Solvent Relaxation
Fluorescence occurs at a lower frequency than the incident radiation because the emissive
transition occurs after some vibrational energy has been released into the surroundings. The vivid
oranges and greens of fluorescent dyes are an everyday manifestation of this effect: they absorb
light in the ultraviolet and blue, and fluoresce in the visible. The proposed mechanism also
suggests that the intensity of the fluorescence ought to depend on the ability of the solvent
(protein environment) to accept the electronic and vibrational quanta, It is indeed found that a
solvent composed of widely spaced vibrational levels (such as water!!) can in some cases accept
the large quantum of electronic energy and so extinguish or quench, the fluorescence.
Fig 6.24The solvent can shift the fluorescence spectrum relative to the absorption
spectrum. On the left we see that the absorption occurs with the solvent molecules (the
ellipses) in the arrangement characteristic of the ground electronic state of the molecule
(the sphere). However, before the fluorescence occurs, the solvent molecules relax into a
new arrangement, and that arrangement is preserved during the subsequent radiative
transition.
- 146 -
The Jablonski Diagram
Jablonski Diagram
Internal conversion: 10
-12
sec
Fluorescence: 10
-8
sec
Intersystem Crossing: 10
-8
sec
S
0
S
1
S
3
S
2
21
21
A k k
A
F
isc ic
+ +
= |
Quenching process
Q isc ic
k A k k
A
F + + +
=
21
21
|
When a molecular system is excited by light into a high vibronic state, there exist multiple
pathways for relaxation. Generally, the fastest process is internal conversion in which the
molecule exchanges vibrational quanta with its environment. For a molecule with a dense
manifold of vibrational states, inside a protein environment also with a dense manifold of
vibrational states, this exchange is easy and occurs with a typical rate of 10
13
-10
12
s
-1
. Note that
during this relaxation fluorescence may occur, but the total amount of fluorescence observed
from these higher vibronic states is very low and difficult to detect.
Typically, the molecule is within 10
-12
s in the lowest excited state, denoted S
1
, implying a
singlet state, in case the ground state was also a singlet. For fluorescent molecules, the decay via
internal conversion from the lowest excited singlet state to the ground state is slow. The main
reason is the large energy gap between S
1
and S
0
often in combination with a rigid molecular
frame. For instance for chlorophyll a the lifetime of S
1
is a few ns. Therefore, from S
1
alternative
decay pathways may be active. Specifically, the spins, which are originally singlet (antiparallel)
may change into triplet (parallel). This process is called intersystem crossing, for chlorophyll a
the rate is about k
isc
=10
8
s
-1
. Also in addition to fluorescence, internal conversion may remain
Fig 6.25 Jablonski diagram summarizing all the possible decay pathways of a molecular
electronic excited state. Typical timescales for the various processes are indicated
- 147 -
active. For chlorophyll internal conversion from S
1
is slow, with a typical rate constant
k
ic
=10
8
s
-1
; in contrast for a DNA base k
ic
=10
12
s
-1
, implying that chlorophyll is rather fluorescent
and DNA-bases are not.
The green fluorescent protein
Probably the most famous fluorescence marker now used in biology is the green fluorescent
protein. Its popularity as a fluorescent marker arises from the fact that it is a small and
exceptionally stable protein, it can be attached using molecular genetics to any protein in the cell,
in almost all cases it does not affect the function
350 400 450 500 550
0
1
B-state
A-state
GFP absorption
a
b
s
o
r
b
a
n
c
e
wavelength (nm)
The structure of GFP
GFP has a |-barrel structure and is
very rigid. The chromophore
originates from three amino acids
that are linked to form an extended
conjugated chain that absorbs in the
blue part of the spectrum
- 148 -
7. The Light-Harvesting Photosynthetic Antenna
7.1 General concept
All chlorophyll-based photosynthetic organisms contain a light-collecting antenna. These
systems function to absorb light and transfer the energy in the form of electronic excitation
energy to a reaction center, where by ultrafast electron transfer the electronic excitation energy is
stored in the chemical free energy of a charge separated state. Other processes compete with the
primary reaction of photosynthesis like fluorescence, internal conversion etc.
AFM view: Bahatyrova, Frese, Siebert, Olsen, van der Werf, van Grondelle,
Niederman, Bullough, Otto and Hunter, Nature 430, (2004), 1058
7.1. Schematic representation of energy transfer among an array of antenna molecules,
symbolized by the green disks. The process continues until a special chlorophyll is reached,
called the Reaction Center chlorophyll where after electronic excitation an electron is
transferred to a neighboring acceptor molecule. Now we know that the pigments are bound to
proteins that form highly organized two-dimensional arrays in the photosynthetic membrane.
The pigment-protein organization in the membrane of the photosynthetic purple bacterium
Rhodobacter sphaeroides was recently visualized by us using AFM.
In contrast to the reaction center complexes (see below) antenna complexes are remarkably
diverse. Here we will first explore some general concepts of antenna systems and then examine
some of the classes of antennas with an emphasis on the structure of antennas in relation to their
energy transfer properties. We finally note that the concept of excitonic interaction between
neighboring pigments, via the electronic coupling of their electron clouds, plays a crucial role in
describing much of the antenna phenomenology.
- 149 -
7.2. A bit of history
One of the first hints that pointed the way to the concept of an antenna came from the
experiments by Emerson and Arnold (1932) who found that one oxygen molecule is produced
for only about 2500 chlorophyll molecules after a saturating flash of light. Although at the time
multiple interpretation were suggested, it were Gaffron and Wohl (1936) who suggested the idea
of a photosynthetic unit in which many chlorophylls cooperate to produce the oxygen. How such
an energy transfer process could occur was unknown at the time, since no physical mechanism
was available. The idea that electronic energy was efficiently transferred was even heavily
contested by James Franck and Edward Teller (1938). It was Frster in 1948 who provided the
physical mechanism (see below). In the early fifties Duysens in Leiden provided the
experimental evidence by careful absorption and fluorescence experiments
7.3. Why antennas?
Why is not every chlorophyll capable of carrying out complete photosynthesis? This would
avoid a lot of complexity and structural organization so typical for the photosynthetic apparatus.
The answer comes from a consideration of the intensity of sunlight in relation to the economics
of cellular processes. Given the intensity and spectrum sunlight and the absorption properties of
a chlorophyll one calculates that in full sunlight every chlorophyll is excited about 10 times per
second. Note that on average this may easily be an order of magnitude lower. Viewed in this way
sunlight is a rather dilute energy source. Let us now compare this with the typical rates of
enzymes. A tenth of a second is an eternity on a molecular scale. If every chlorophyll had
associated with it the entire electron transfer chain and enzymatic complement to finish the job
of photosynthesis, then these expensive components would sit idle most of the time, only
springing into action when a coincidental photon is absorbed. So the cooperation between
chlorophylls to deliver their excitation energy at the reaction center is like the cooperation
between farmers who all deliver their milk at the milk factory. One reaction center per few
hundred chlorophylls is able to turnover at a maximum rate. One additional aspect is that many
of the photosynthetic reactions require the action of more than one photon. For instance to
oxidize water and produce molecular oxygen in Photosystem 2 of plants requires 4 charge
separation events and all the intermediates are unstable. To avoid loosing these unstable
intermediates demands that photons be collected and their energy delivered to and used in a
central place.
7.4. Classes of antennas
A remarkable variety of antennas is found in various photosynthetic organisms. In fact, in many
cases, the groups of photosynthetic organisms are largely defined by the types of antennas they
contain. There are a number of major classes of antennas, which show no apparent relation to
each other in terms of structure or even types of pigments utilized. This suggests that there have
been several independent evolutionary origins of antennas, but it also underscores the importance
of a light-harvesting system.
Almost all antenna complex are pigment-proteins in which the chlorophyll or other pigment is
specifically associated with proteins in a unique structure. The only known exception to this rule
- 150 -
is the chlorosome antenna complex, containing 1000s of self-assembled (through pigment-
pigment interactions) bacteriochlorophyll c molecules, and which functions as a light-harvesting
antenna in green photosynthetic bacteria (see below).
Antenna complexes can be broadly divided into integral membrane antenna complexes and
peripheral membrane antenna complexes. Integral membrane antenna complexes are
composed of proteins that cross the membrane. In peripheral antenna complexes the antenna
complex is associated with components buried in the membrane, but does not itself span the
membrane. Such a complex is invariably attached to just one particular side of the photosynthetic
membrane. Energy absorbed by the pigments in the peripheral antenna complex is transferred
into the integral membrane antenna complex and eventually into the RC where photochemistry
takes place.
The integral membrane antenna complexes are themselves quite diverse in terms of structure and
relative position in the energy transfer sequence. Some of these antenna pigments and their
corresponding antenna functions are actually built into the minimal reaction center structure.
These pigments can not be separated biochemically from the electron transfer components,
because they are bound to the same polypeptides. We will call the systems fused
antenna/reaction center complexes. The best example is the Photosystem 1 complex, the best
understood example of such a fused system.
1
The Photosystem I reaction center has 12 subunits
It binds about 100
molecules of Chl. Those
shown in yellow are part
of the antenna.
The arrangement of the Chl electron
carriers (blue) again is very similar
to that in bacterial RCs.
view normal to
the membrane
Fig 7.2. Top view of the Photosystem 1 core complex. The Photosystem 1 core complex is an
integral membrane protein consisting of 12 protein subunits. It binds about 100 chlorophyll
molecules, 6 of which (blue) are positioned in the center of the complex and perform
transmembrane charge separation while the other 94 chlorophylls (in yellow) and 22 |-
carotenes (in black) function as antenna pigments, absorbing the light and transporting the
energy to the point where the electronic excitation is used to perform a charge separation.
- 151 -
The second class of integral membrane antenna complexes are the core antenna complexes.
These antennas are closely associated with the RC, occur in some fixed arrangement and
stoichiometry, but can in principle be biochemically separated. Good examples are the purple
bacterial RC-LH1 core (see fig 7.3) and the PS2 core
1
The antenna system of purple photosynthetic
bacteria has circular pigment-protein complexes
view normal to the membrane
The LH1 complex, with 31 transmembrane
o-helical peptides (yellow) and 30 BChls
(CPK colors), surrounds the reaction
center (red, orange & brown), where the
electron-transfer reactions occur.
view in the plane of the membrane
A. W. Roszak et al. Science 302: 1969 (2003); 1pyh.pdb
Fig.7.3. Structure of the bacterial RC-LH1 core complex. The LH1antenna surrounds the RC
and is composed of two concentric rings of small transmembrane, o-helical polypeptides (|-
peptide outside, opeptide inside) with a ring of 30 BChls sandwiched between them. The total
diameter of the complex is about 10 nm, the center-to-center distance between two neighboring
BChls in the ring is less than 1 nm, which makes them strongly coupled. Note the hole in the
ring, which allows quinones to diffuse in (Q) and out (QH
2
) of the RC-LH1 complex.
The last group of integral membrane antenna complexes are called accessory antennas, to
emphasize the fact that they are always found in addition to the core, or fused antennas, not in
place of them. They are often present in variable amounts, depending on growth (and light)
conditions, and maybe mobile, in that their physical arrangement with the other antennas and
RCs is not fixed in either time or space. They are often involved in antenna regulatory processes
(like the distribution of excitation energy between photosystem 1 and 2 in plants and green
- 152 -
algae). Important examples are the LH2 antenna complex that in (some) photosynthetic purple
bacteria feeds excitations into the LH1-RC core, Lhc2 in plants, the most abundant protein on
earth, that is mostly associated to Photosystem 2 (see Fig.7.4) and Lhc1 that is connected to
Photosystem 1.
1
Top-view of the dimeric
Photosystem II supercomplex.
L, S, M are accessory Lhc2
trimers
Top-view of the Lhc2
trimer crystal structure
Chl a is blue, Chl b is green,
carotenoids orange. There are 8
Chl a, 6 Chl b and 4 carotenoids
in each monomer
Fig.7.4 Left: topview of the pigment-protein arrangement of the dimeric Photosystem2
supercomplex. In the center a dimer of Photosystem 2 cores (D1,D2,CP47,CP43, blue)
connected via the minor Photosystem2 antenna complexes CP24, CP26 and CP29(red) to
trimers of Lhc2 (L,M.S, green). Right: topview of the Lhc2 trimer of spinach, for which a crystal
structure was obtained. The trimer contains 24 Chl a, 18 Chl b, 12 carotenoids (6 luteins, 3
neoxanthins, 3 violaxanthins).
7.5. Physical Principles of Antenna Function
In this section we will discuss some of the physical measurements and mechanism that are
important in studies of antennas. We will begin with some organizational concepts, then take up
experimental aspects of how antennae are investigated and finally describe shortly the basic
theory of energy transfer and pigment interactions. A more extensive discussion of excitons and
energy transfer is given in the next chapter.
1 The funnel concept
The analogy of an energy funnel is a useful image for visualizing the energy collection in
antenna systems. This is illustrated in fig. 7.5.
- 153 -
The more distal parts of the antenna, often a peripheral antenna complex such as a
phycobilisome, maximally absorbs photons at shorter wavelengths than do the pigments in the
antenna complexes that are proximal to the reaction center. Since net energy transfer will occur
from pigments absorbing at shorter wavelength to those absorbing at longer wavelength, energy
will flow from the periphery to the pigments physically close to the RC. With each transfer event
a small amount of energy is lost as heat, and the excitation is moved closer to the RC. The
energy lost in each step provides a degree of irreversibility to the process, so the net result is
that the excitation is funneled into the RC where the energy is eventually stored by
photochemistry.
It is not absolutely necessary that each individual energy transfer event be downhill
energetically. At room temperature thermal energy is always available to add to the excitation
energy to increase it somewhat, so occasional transfers can be from lower-energy excited states
to form higher-energy ones. However, the probability of these uphill energy transfers becomes
exponentially smaller as the energy gap increases, so that, in the end, the excitations will be
delivered from the higher-energy pigments to the lower-energy ones.
The funnel concept can be viewed as a mechanism in which a portion of the photon energy is
sacrificed as heat to deliver the energy in a short time to the proximity of the reaction center trap.
A large antenna system that is isoenergetic with the trap might absorb energy equally well, but
much of it might be lost if the excitations had to wander around the antenna before eventually
finding their way to the RC. For the funnel arrangement to work, there must be both spatial and
spectral ordering of the pigments, so that the blue pigments are farthest away from the trap and
the longest-wavelength absorbing ones are nearest to the trap. In many cases, this arrangement is
Fig 7.5 The funnel concept. The photosynthetic light-harvesting antenna is organized in
such a way that blue absorbing pigment-proteins are far away from the reaction center
and red absorbing pigments close by. In this way the excitation energy is concentrated
close to the RC.
- 154 -
realized by nature; a good example is the phycobilisome, the peripheral antenna of
cyanobacteria.
In a slightly different form the funnel concept also applies within an antenna complex. Accessory
pigments, most commonly carotenoids, absorb intensely at short (400-500 nm) wavelengths.
These accessory pigments provide a much broader coverage of the solar spectrum than is
possible with just chlorophylls (note that the peak of the solar emission is around 500 nm, where
chlorophyll hardly absorbs). Energy absorbed by these accessory pigments is rapidly transferred
to chlorophylls within the same antenna complex, and thus available to drive photosynthesis.
The funnel concept seems to be most applicable in cases of peripheral membrane antenna
complexes. For antenna complexes that directly interact with the reaction center complex, either
the integral membrane core antennas or the fused antennas, the funnel model breaks down. In
these situations, the absorption of the complex occurs within a rather narrow band (for instance
for the Chls a of the PS2 and PS1 cores in the region 665-680 nm, for the BChls a in LH1 around
870-880 nm). Some antenna pigments even absorb at an energy significantly lower than that of
the trap, so an uphill energy transfer step is needed before the energy can be used. The functional
significance of these low energy antenna pigments is most likely to increase the spectral
coverage at the expense of a slightly slower trapping process.
2 Antenna organization
The concept of the photosynthetic unit emerged from the Emerson and Arnold experiment and its
subsequent analysis. However, the details of how the antenna pigments and reaction centers are
organized in the photosynthetic membrane was for a ogn time unknown. Only recently electron
micrographs and AFM images have become available for some photosynthetic membranes.
Some specific arrangements are shown in Figs 7.6, 7.7 and 7.8.
AFM-view
of a native photosynthetic membrane
Bahatyrova et. al. Nature 2004
- 155 -
Fig.7.6 Organization of the photosynthetic membrane of Rhodobacter sphaeroides as detected
with AFM. Note the rows of dimers of RC-LH1 (large rings with RC in the center) surrounded by
domains of LH2 (small rings)
1
Supramolecular organization
in photosynthetic membranes
Electron microscopy (UG)
Spectroscopy (VU)
Fig 7.7. Left top Stacked(green) and unstacked (blue) regions of the thylakoid membrane. In the
stacked regions or grana one finds Photosystem 2, in the unstacked regions or stroma
Photosystem 1 is located. Bottom left: Time-resolved fluorescence emission experiment on
isolated Photosystem1 particles. Fluorescence spectra have been measured as a function of time
using a femtosecond laser pulse to excite the fluorescence and a streak camera to detect the
fluorescence. Note that there is a 3.4 ps component that characterizes blue to red energy
transfer in Photosystem 1. There are decay components in the 50-500 ps time range that can be
identified as trapping from the core and the surrounding Lhc1 antenna. Note that the lifetimes
are all much shorter that the fluorescence lifetime of chlorophyll in solution of about 5 ns.
Center: The Photosystem 1 structure. The Photosystem 1 core is surrounded by a variable
number of Lhc1 (accessory antenna). Top right: Electron microscopy on single photosynthetic
particles. Image analysis yields low resolution (1-2 nm) structures, like those shown on the
bottom right for the IsiA complex in various states of appearance. The IsiA antenna protein is
produced by cyanobacteria under iron stress, as a strategy for survival. IsiA has a strong
tendency to form rings. In the center is a trimer of Photosystem1 cores surrounded by a ring of
IsiA; the IsiA ring transfers excitation energy to the Photosystem core trimer in the center; to
the left is a single IsiA ring, to the right a double ring.
- 156 -
1
About 200 chlorophylls per photoactive pigment
Fig7.8. Topology of the grana membrane reconstructed from electron microcopy images. Rows
of dimeric Photosystem 2 supercomplexes are observed, similar to those shown in fig7.4.
C=Photosystem2 core, M,S are Lhc2 trimers. Note how this arrangement is actually similar to
that obtained with AFM for photosynthetic purple bacteria.
In figs 7.6-7.8 a variety of antenna arrangements is shown. One thing that is striking is the high
degree of connectivity of the photosynthetic light-harvesting apparatus, explaining (at least
structurally) the amazing efficiency of the antenna function. One conspicuous property is that the
antenna is not composed of discrete units, each serving just one RC, but that excitations
delivered from the peripheral antenna complexes still may travel to more than one RC. This is of
course advantageous under conditions of high light, where one RC may be busy, closed and
consequently can not deal with the next excitation. Then the excitation can travel further to an
open RC. This has a very interesting consequence for how the fluorescence yield of such a
complex system depends on the fraction of closed and open reaction centers. This can be
(relatively) easily measured by monitoring the fluorescence and the fraction of open/closed RCs
(by absorbance difference spectroscopy) at the same time (see below)
- 157 -
7.6 How to measure antenna function.
1. Fluorescence quantum yield
A measurement of the fluorescence quantum yield is an easy measurement of the efficiency of
light-harvesting. During the energy transfer process there is a small, but finite probability that the
energy is lost as fluorescence. The quantum yield of chlorophyll fluorescence in a solvent is
between 20-30%. The quantum yield of chlorophyll fluorescence in a chloroplast is about 0.5%,
implying an efficiency of photosynthesis of about 97%. However most of this fluorescence
originates from photosystem 2, which is obviously the least efficient photosystem. Fig. 7.8
suggested, on the basis of electron-microscopy imaging, that the Photosystem 2 reaction centers
are all connected to one large light-harvesting antenna. One way to probe this connectivity of
the photosynthetic apparatus is to measure the fluorescence quantum yield as a function of the
number of busy or closed reaction centers. Typically, when a chloroplast is illuminated the
fluorescence starts low (all the traps are active), but then rapidly increases with time because
traps are photochemically closed. For Photosystem 2 this closed state is the state where the first
stable electron acceptor, a quinone, is reduced and the formation of a stable charge separation is
no longer possible. Under those conditions the trap works at a much lower rate and the
fluorescence is high. In chloroplasts one may easily measure a factor 4-5 increase in fluorescence
yield upon closing the traps.
Suppose the arrangement of Photosystem 2 in the membrane is as shown in fig. 7.8. And suppose
that energy transfer is so efficient, that at any moment in time we can consider the electronic
excitation as equilibrated, meaning that the probability to find an excitation at a certain site is
independent of the postion in the lattice. Then, with a fraction 1-q of open reaction centers,
trapping at a rate
open
k and a fraction q of closed reaction, trapping at a rate
closed
k the amount of
excited states is given by:
( ) 0 * * 1 *
*
= = Chl qk Chl k q Chl k cIChl
dt
dChl
closed open l
Where c is a constant, I is the light intensity, Chl is the concentration of not excited chlorophylls,
Chl* is the concentration of excited chlorophylls and
l
k is the rate of loss of the excited state.
Furthermore, we have assumed a steady state or 0
*
=
dt
dChl
.
Then the fluorescence yield as a function of q is:
( )
( )
closed open l
f f
fl
qk k q k
k
cIChl
Chl k
q
+ +
= =
1
*
| (7.1)
With
f
k the rate of fluorescence.
- 158 -
This is easily rewritten as
( ) ( )
pq
q
fl fl
=
1
1 1
| |
with ( )
f closed open
k k k p = (7.2)
Eq. 7.2 predicts a hyperbolic relation between the fluorescence quantum yield and the fraction of
closed traps. This relation was indeed experimentally observed, long before detailed images like
in fig 7.8 were available, and interpreted in the way discussed above, namely that in particular
Photosystem 2 is a highly connected network of antennas and RCs.
2. Excitation spectra
The second method of experimentally establishing the process of energy transfer is to measure
fluorescence excitation spectra. The idea is that if A and B are connected via energy transfer,
then exciting A will result in the fluorescence of B. Therefore, if we measure the fluorescence
excitation spectrum of B, we should find a contribution of A. How much A we observe
determines the energy transfer efficiency. This method was employed for the first time by
Duysens to establish the energy transfer from carotenoids to bacteriochlorophyll in purple
photosynthetic bacteria and the transfer from chlorophyll b to chlorophyll a in plants. The
method is illustrated in fig 7.9.
Fig 7.9. Energy transfer efficiency from fluorescence excitation measurements. A schematic
picture of absorbing (A) and emitting (B) molecules with energy transfer between them is
shown at the top. In a fluorescence excitation experiment one scans the wavelength of the
exciting light and detects the amount of fluorescence. This is than compared with a 1-
transmission (1-T) spectrum of the same material
- 159 -
Fig 7.10 gives on example of how fluorescence excitation spectroscopy can be used to map the
energy transfer pathway of a complex light-harvesting system. Chloroflexus aurantiacus is a
green photosynthetic bacterium that has both a membrane bound light-harvesting system,
surrounding the RC, like in fig 7.3. and a large accessory antenna called the chlorosome,
containing a few 1000s of Bacteriochlorophyll c, that are self-assembled into a large aggregate,
which is surrounded by a membrane. This whole structure is called a chlorosome, it is attached to
the photosynthetic membrane, and excitations are transferred from this chlorosome to the
membrane-bound RC-LH1 antenna with a high, but not 100% efficiency.
A schematic representation of the photosynthetic membrane of the green sulfur bacterium
Chlorobium tepidum is shown in fig. 7.11. The large structure filled with BChl c aggregates
attached to the membrane is the chlorosome
Fig 7.10 Measurement of energy transfer efficiency in whole cells of the green bacterium
Chloroflexus aurantiacus. Solid line: 1-T spectrum; dotted line: fluorescence excitation
spectrum with the fluorescence monitored at 900 nm. Dashed line: fluorescence emission
spectrum. The energy transfer efficiency of transfer from the bacteriochlorophyll c
(chlorosomes) absorbing at 740 nm to the bacteriochlorophyll a emitting at 900 nm is about
70%, while the transfer efficiency for bacteriochlorophyll a absorbing at 808 nm is 100%
- 160 -
3. Time-resolved fluorescence spectroscopy
Time-resolved fluorescence spectroscopy is probably the most adequate method to reveal energy
transfer pathways. Following selective excitation of a specific group of pigments one can follow
the fate of the excitation with high time (<0.1ps) and high spectral resolution (note that the
spectral resolution is inversely related to the time resolution via the uncertainty relation).
An example of such an experiment is shown in fig 7.12
Fig 7.11 A schematic representation of the photosynthetic membrane of the green
sulfur bacterium Chlorobium tepidum. The large structure filled with BChl c
aggregates and surrounded by a single membrane attached to the membrane is the
chlorosome
- 161 -
Streak camera experiment on Photosystem 1 complex
wavelength
t
i
m
e
4. Multi-photon spectroscopy
In multiphoton spectroscopy one employs the fact that in a large connected system like that of
Photosystem 2 it is easy to generate two (or more) excitations at the same time. Since both
excitations move around at a high speed, they may meet and in such a case they may annihilate.
What happens is that upon collision a chlorophyll becomes excited to a higher excited state,
which then decays rapidly (in less than 100 fs) to the lowest excited state, and the energy of one
of the two excitations is lost as heat (see fig 7.13).
Fig 7.12. Photosystem 1 of the cyanobacterium Synechocystis sp. was excited in at 400 nm and
the fluorescence was detected with a streak camera in the wavelength region 620-775 nm. In the
center the structure of Photosystem 1 is shown (a, topview; b, side view). There are 100
chlorophylls in photosystem 1, whose positions are known with an accuracy of less than 0.3 nm.
Left: image of the streak camera in false colors. Right: Fluorescence kinetics and decay
associated spectra resulting from a model analysis of these data. In early times (up to 10 ps)
there is a lot of spectral dynamics (note the components of 0.8 and 4.4 ps), reflecting the energy
transfer among the chlorophyll a pigments in Photosystem 1. Most of the excitations are trapped
on a time scale of about 24 ps.
- 162 -
Energy transfer in nano-engineered ensembles of photosynthetic light-harvesting complexes: towards a bio-solar cell
Multi-Excitation Annihilation
Energy Transfer Annihilation
What one observes is that with increasing intensity of the exciting laser pulse the fluorescence
gets quenched. The point at which the quenching starts depends on two parameters: the number
of connected pigments and the (average) rate of energy transfer among the pigments.
Fig 7.13 Scheme or multi-photon annihilation. In a network of pigments, connected via
fast energy transfer there are two excitations simultaneously present
- 163 -
8. Excitons and Excitation Energy Transfer.
8.1. Introduction
To harvest solar photons, photosynthetic organisms are equipped with a light-harvesting antenna.
Antenna pigment-proteins contain many pigments, often at distances of less than 1 nm, and as a
consequence the coupling between local excited states becomes a crucial factor in describing
their spectroscopy and energy transfer function. This coupling between localized excited states is
often referred to as exciton interaction.
8.2. The Excitonically Coupled Dimer
8.2.1 The exciton states
The simplest system for which we can consider exciton effects is a pair of interacting molecules.
We will call such a pair an excitonically coupled dimer, where it should be noted that the word
dimer does not imply that the two molecules are in Van der Waals contact., in fact they can be
spatially separated. First we consider two molecules in a medium with dielectric constant c ,
separated at a fixed distance r
12
having a fixed orientation in space and each having only two
energy levels (0,1). For a given Hamiltonian H the isolated molecules have their two eigenstates
i
n
determined by:
i
n
i
n
i
n n
E H =
1 , 0 ; 2 , 1 = = i n
(8.1)
where the subscript n identifies the pigments (1,2) and the superscript i refers to the ground (0)
and excited state (1). The ground state of the non-interacting dimer is
0
2
0
1
; there is a
degenerate pair of single excited states
0
2
1
1
and
1
2
0
1
.
When the two molecules interact (via Coulombic interaction) the total Hamiltonian includes
besides H
1
and H
2
also the interaction V between them. In that case
0
2
1
1
and
1
2
0
1
are no
longer the correct excited states of the system and also the corresponding eigen energies will
have changed giving rise to perturbations of the absorption spectrum. In case V is weak, we can
use perturbation theory for degenerate states with the Heitler-London approximation, which
implies that the eigenfunctions of the dimer are equal to linear combinations of the product of
molecular eigenfunctions. In first order the ground state remains
0
2
0
1
0
= with the
corresponding ground state energy of the coupled dimer given by:
00
0
2
0
1
0
2
0
1
0
2
0
1
0
2
0
1
0
2
0
1 2 1
0
2
0
1 0
V E E V E E V H H E + + = + + = + + = (8.2)
- 164 -
where the last term is a Van der Waals interaction between the permanent dipoles of the two
molecules. In general the ground state energy of the dimer is lowered by an amount
00
V relative
to the summed ground state energies of the monomers.
The excited states can be written as:
1
2
0
1 2
0
2
1
1 1
f f
f
c c + =
(8.3)
The new excited state wavefunctions must be normalized and orthogonal, so the coefficients
1 f
c
and
2 f
c must fulfill the following relations:
0
1
2
*
2 1
*
1
2
2
2
1
= +
= +
g f g f
f f
c c c c
c c
(8.4)
where f and g represent different eigenstates. Thus the excited state f of the coupled system is a
linear combination of two terms in which either one or the other molecule of the dimer is
excited. The relative contributions of these two terms are determined by the coefficients
2 , 1 f
c .
The new eigenstates are required to be stationary solutions of the Schrdinger eq. of the dimer,
thus:
( )
f
D
f
D
f
D
E V H H = + +
2 1
2 , 1 = f
(8.5)
Multiplying both sides from the left with either
0
2
1
1
or
1
2
0
1
and integrating over space yields
the two following equations:
( )
( )
f
D f f f
f
D f f f
E c V E E c V c
E c V c V E E c
2
1
2
0
1
1
2
0
1
1
2
0
1 2
0
2
1
1
1
2
0
1 1
1
1
2
0
1
0
2
1
1 2
0
2
1
1
0
2
1
1
0
2
1
1 1
= + + +
= + + +
(8.6)
which can be written in a shorthand notation as
( )
( ) 0
0
22
1
2
0
1 2 21 1
12 2 11
0
2
1
1 1
= + + +
= + + +
f
D f f
f
f
D f
E V E E c V c
V c E V E E c
(8.7)
Here
12
V and
21
V are the resonance interaction terms, which are also the quantities that determine
the transfer of excitation energy between the molecules 1 and 2, as will be discussed later. This
can already be anticipated from the explicit form of eq 8.6, where in the bra term one molecule
is excited whereas in the ket term the other molecule is excited. Non-trivial solutions to 8.7 are
obtained if:
- 165 -
0
22
1
2
0
1 21
12 11
0
2
1
1
=
+ +
+ +
f
D
f
D
E V E E V
V E V E E
(8.8)
Assuming the two molecules to have identical site energies (which is generally not the case) we
have: 0
0
2
0
1
= = E E ,
1 1
2
1
1
E E E = = ,
21 12
V V = , and
22 11
V V = , and then it follows that
( )
2
12
2
11
1
V E V E
f
D
= + 2 , 1 = f (8,9)
leading to two eigen energies
2 , 1
E given by:
12 11
1 2
12 11
1 1
V V E E
V V E E
D
D
+ =
+ + =
(8.10)
The excitonically coupled dimer
The energy levels have been split by an amount 2V
12
(Davydov or
exciton splitting. The average of these two levels has shifted by an
amount V
11
-V
00
(also called the displacement energy D). Usually a
redshift.
Substituting E
1,2
into the secular equations yields:
( )( )
1
2
0
1
0
2
1
1
1
2 1 + =
and ( )( )
1
2
0
1
0
2
1
1
2
2 1 =
In both states the excitation is fully delocalized over both molecules.
monomer
dimer
V
11
V
00
2V
12
E
1
E
2
Fig 8.1. Exciton splitting. Note that V
12
is not necessarily positive. The sign depends on the
geometry of the dimer (see below).
From 8.10 it follows that the energy difference between ground- and excited state (the transition
energy) has changed as compared to that for a (Chlorophyll) monomer. The energy levels have
split by an amount
12
2V (Davydov or exciton splitting) and the average energy of these two levels
has been shifted with respect to the ground state by an amount of
00 11
V V , which is the so-
called displacement energy D. In fact D is comparable to the change in energy that a molecule
- 166 -
experiences when it goes from the gas phase to a solution. This usually leads to a red shift of the
band. Since this term is not important for our further discussion about excitons we will discard it
in the following. The value of the resonance interaction can then be calculated from the
intermolecular interactions (basically the dipole-dipole interaction) and we will return to this
point below.
For ease of presentation we have set
0
E equal to 0. As long as this term is the same for all
molecules this is allowed since it only leads to a displacement of all the energies. If it is not equal
for all molecules, we can still account for this difference by including the deviation into the
values of
1
1
E and
2
1
E . Variations in site energies occur for instance pigments are located at
different positions in a protein (see below). We finally note that the level ordering as indicated in
fig.8.1 is not necessarily the correct one, since we do not know the sign of V. Since V is a dipole-
dipole interaction term it may be positive or negative, depending on the geometry of the system.
Substituting the resulting values for
2 , 1
E into 8.7 leads to the following excitonic wavefunctions
( )
1
2
0
1
0
2
1
1
1
2
1
+ = and ( )
1
2
0
1
0
2
1
1
2
2
1
= (8.11)
Note that in both excited states the excitation is fully delocalized over both pigments.
1
and
2
are also called the in phase and out of phase combinations of the locally excited states
0
2
1
1
and
1
2
0
1
. There is a nice analogy with two pendulums coupled by a weak spring.
8.2.2 The redistribution of oscillator strength
The transition dipole
0
1
1
1
01
= is a measure for the interaction between the charge
density of molecule 1 and an oscillating electrical field. Idem for molecule 2. The dipole strength
is a measure for the intensity of the absorption/emission of that specific transition: ( )
2
01
01
= D .
01
D is calculated from the absorption spectrum via
( )
e
e
e c
d D
band
}
=
3
01
10 18 . 9 , where
01
D is obtained in Debye
2
.
For the excitonically coupled dimer the two excitonic transitions acquire a different dipole
strength, dependent on the structure of the dimer.
( )( ) ( ) u cos 1 2 1
01
2
0
2
0
1 2 1
1
2
0
1
0
2
1
1
2 , 1
= + = D D
(8.12)
- 167 -
Where u is the angle between the two monomeric transition dipoles. For instance, if 1 cos = u ,
0
1
= D , i.e. no absorption occurs to the level belonging to the + state, while at the same time
01
2
2D D = , i.e. all the oscillator strength is accumulated in the transition to the level belonging
to the - state (not necessarily the lowest in energy; see below).
8.2.3 The coupling matrix element
The electrostatic interaction energy between two uncharged molecules with a relative position
vector
j i
r r R
= is, in first order. given by the dipole-dipole interaction:
( )( )
=
3
. 3 .
4
1
R
R R
V
i j i j
tc
(8.13)
Note that when the two molecules get closer than say 2 nm higher order terms in the electrostatic
interaction become important.
Then
( )( ) ( )( )
3
01
2
01
1
01
2
01
1 1
2
0
1 3
2 1 2 1 0
2
1
1 12
3
3
R
R R
R
R R
V
=
=
(8.14)
This is a nice result because we can now calculate the interaction strength on the basis of our
knowledge of the transition dipoles and the geometry of the dimer.
Frequently the coupling strength is written in short-hand form as
3
01
12
04 . 5
R
D C
V
k
= with ( ) ( ) ( ) R R
, cos , cos 3 , cos
2
01 01
01
2
01
1
k = (8.15)
Where
r l
f C c
2
= , where
l
f is the Lorentz factor. With Din debye
2
and R in nm,
12
V is obtained in cm
-1
.
- 168 -
The interaction matrix element
3
01
12
04 . 5
R
D
C V
k
=
with ( ) ( ) ( ) R R
, cos , cos 3 , cos
01
2
01
1
01
2
01
1
k =
and
r
l
f
C
c
2
= , where f
l
is the Lorentz factor. With D in debye and R
in nm, V
12
is obtained in cm
-1
.
head-to-tail
sandwich
3
01 12
2 R D V =
3
01 12
R D V =
+
-
red-shifted
superradiant
+
-
blue-shifted
dark
Fig 8.2 Interaction matrix element, excitonic energy levels, and corresponding dipole strengths
for a head-to-tail and sandwich dimer.
8.5 The disordered dimer
Proteins are in principle disordered in the sense that although they have a well-defined average
structure, if you would overlay the structures of many individuals at a certain moment in time
significant fluctuations around this average structure would be observable. This phenomenon
(called disorder) is illustrated in fig.8.3 where a set of structures of the protein chymotrypsin are
shown as they occur at various points in time during a molecular dynamics simulation. As a
consequence also the pigments in a protein environment do not necessarily have identical
environments and this causes that each pigment has its own transition frequency, the distribution
of transition frequencies is approximated by a Gaussian distribution (the inhomogeneous
distribution function or IDF)
- 169 -
The Disordered Dimer(1)
Proteins are intrinsically
disordered. There exist many
energetically equivalent states
Energy
Select site energies from
am Inhomogeneous
Distribution Function
(IDF)
P(E)
The spectral properties of a disordered dimer we can now calculated by diagonalizing the
Hamiltonian
2
1
E V
V E
H = for a large number of combinations of
2 1
, E E , where
2 1
, E E are
randomly taken from the inhomogeneous distribution function. For each choice the spectral
properties of the dimer are calculated, they are added and eventually the ensemble spectrum of
the disordered dimer is obtained. What is funny is that the calculated spectrum now has noise,
because the calculation has usually been done for say a few 1000 of
2 1
, E E combinations.
8.6 Energy transfer within the dimer.
The coupling matrix element
1
1
0
2
0
1
1
2 12
V V = physically implies the transfer of the
excitation between the molecules 1 and 2. On what timescale does this transfer occur? To
estimate this we have to look at the time-dependent Schrdinger equation for the dimer.
For a general non-stationary state ( ) t we have: ( ) t i t H c c = with
Fig 8.3 Disorder. The top figure shows an overlay of a number of backbone conformations
of the protein chymotrypsin as they occur during a molecular dynamics simulation.
Although all structures resemble the average, there are clear differences between the
different backbone conformations. The spectra of pigments bound to proteins are sensitive
to sub-nm changes in distances and small reorientations of the residues that surround them.
As a consequence their exists a distribution of site energies (the inhomogeneous
distribution function, IDF) often approximated by a Gaussian.
- 170 -
( ) ( ) | | ( ) | | t E i c t E i c t
D D
2 2
2
1 1
1
exp exp + = (8.16)
With
2 1
, E E given by 8.10 and
2 1
, by eq.8.11. Assuming that at 0 = t molecule 1 is excited
and 2 not, then eq.8.16 represents an oscillation of the excitation between the two molecules.
( )
(
|
.
|
\
|
|
.
|
\
|
=
1
2
0
1
12 0
2
1
1
12
1
sin cos exp
t V
i
t V
t E
i
t (8.17)
One can define a quasi energy transfer rate as the inverse of the amount of time it takes to find
the maximum excitation density on pigment 2 after it started on pigment 1. Then
h V W
12 12
4 = (8.18)
Note: at all times there is a perfect phase relation between
0
2
1
1
and
1
2
0
1
.
In this strongly coupled regime, when the system is in a certain exciton state the excitation
oscillates between the two halves of the dimer. Furthermore, relaxation between different
exciton states can occur, this is described by a theory for relaxation, implying the interaction
between a quantum system and the outside world (the bath) which is beyond this lecture series.
When the coupling is weak (weak relative to this exciton-bath coupling) then we can apply
Fermis Golden Rule to calculate the energy transfer rate. This will lead to the famous Frster
equation for energy transfer between a donor and an acceptor.
8.7 The Frster Equation
For weakly coupled systems the rate of energy transfer from a donor to an acceptor is given by
Fermis Golden Rule
( ) E V
h
W
DA DA
t
2
2
4
=
(8.19)
Where
DA
V is the (weak) electronic coupling between D and A and ( ) E is the density of states
(meaning the number of combinations D,A where the energy can be exchanged with energy
conservation).
The coupling matrix element is given by the dipole-dipole coupling
6 2
2 2
2 4
R
f V
r
A D
DA
c
k =
and
A D
u k cos cos 3 cos =
(8.20)
The density of states is illustrated in fig.8.4
- 171 -
The Frster Equation (1)
Absorption of A
Emission of D V
( ) E
When we realize that ( ) E consists of all the possible combinations as indicated in fig 8.4, we
can rewrite 8.19 as:
( ) ( ) | | ( ) ( ) | | v v v
c
k t
d dE h E E S E g dE h E E S E g
R
f
W
A A A A A A D D D D D D
r
DA
} } }
+ = , , '
4
2 2 ' ' ' 2 ' 2
6 2 2
2 2 4
(8.21)
Where
2 2
,
A D
S S are Franck-Condon factors for emission of D and absorption of A, resp., and
( ) ) ( ,
' '
A g E g
D
are the Boltzmann factors for excited D and ground A states. The first term of 8.21
is proportional to the emission spectrum of D
( ) ( ) ( )dE h E E S E g n f
c
F
D
R
v v
t t
v =
}
,
3
64
2 2 3 2
3
4
(8.22)
Fig 8.4. The density of states ( ) E measures the number of combinations of states D*A and
DA* that are available for exchange of the excitation energy and conserve the energy. This
obviously corresponds to an overlap between the emission spectrum of D and the absorption
spectrum of A.
- 172 -
Where
2 3 2
3
4
3
64 1
nf
c
k
D R
R
v
t
t
= = is the radiative rate of D. (Note that ( ) v v d F
}
is now
normalized to 1 on a frequency scale. In 8.21 we also recognize the absorption spectrum of the
acceptor A
( )
( )
( ) ( )dE h E E S E g
n
f
c
N
A
av
v
t
v c +
|
|
.
|
\
|
=
}
,
10 ln 10 . 3
8
2
2
2
3
3
(8.23)
Substituting 8.22 and 8.23 we obtain the Frster equation
( ) ( )
v
v
v v c k
~
~
~ ~
10 8 . 8
4 6
2
4
17
d
F
R n
k
W
D A
D
R
DA
}
=
(8.24)
With R in nm and v
~
in cm
-1
.
The Frster Equation (2)
( ) ( )
}
=
4 6
2
4
17
~
~ ~ ~
10 8 . 8
v
v v v c k d F
R n
k
W
D A
D
r
DA
Frster Equation
D abs
D emission
A Abs.
A abs
Excellent Overlap
Bad Overlap
Overlap Integral
The Frster eq. is often written in the following form:
6
0
|
.
|
\
|
=
R
R
k W
D
R DA
with
( ) ( )
}
=
4
17 6
0
~
~ ~ ~
10 9 . 8
v
v v v c d F
R
D A
(8.25)
Fig. 8.5 Illustrating the overlap term in the Frster equation. For fast energy
transfer the emission spectrum of the donor must have optimal overlap with the
absorption spectrum of the acceptor
- 173 -
For chlorophyll a to chlorophyll a energy transfer R is about 1 nm, R0 is typically 8 nm,
1 7
10 5
~ s k
D
R
->
1 13
10
~ s W
DA
8.8 Hopping vs. The Disordered Exciton Model and Redfield
Relaxation
When V is small the excitation hops from chlorophyll to chlorophyll. On every site the coupling
to the bath is so strong that the electronic excitation is fully equilibrated and there is no phase
correlation between the excitation on before and after the hop so the hopping is described by
the Frster equation.. In case V is large we have to find new eigenstates for the system, like we
did for the dimer, but now for a complex with many chlorophylls, we obtain new eigenstates of
the form
n
N
n
nk k
c
=
=
1
, where the
n
are the locally excited states, a new set of energies that
are spread more than the original set of energies (like in the dimer) and the oscillator strength is
redistributed. In each eigenstate many local excited states participate (in principle all) and the
excitation moves coherently among them.
Excitonic Interactions
V
Excitonically
coupledsystem
5
1 2 3 4 5
Etc.
I ndividual pigments
Each with its own Excited State
Wavefunction
Frster ResonanceEnergy Transfer
among Localized states
4
n
N
n
nk k
c
=
=
1
Delocalized states
V small
large
Fig. 8.6.Weak vs. strong coupling
- 174 -
In case a higher exciton state is occupied, relaxation to a lower state can occur by exchanging
energy with the bath (see fig 8.7). This coupling to the bath is represented by a spectral density
( ) e
kp
J
, which reflects the coupling between the exciton states k and p and the bath and tells
you which frequencies are available in the bath to relax. Furthermore, relaxation between exciton
states can only occur if the initial and final state overlap in space, as illustrated in fig. 8.7.
Relaxation In An Exciton Manifold
n
N
n
nk k
c
=
=
1
J
kp
(e)
Delocalized excited state
V
large
Fig.8.7 Relaxation within an exciton manifold. To relax from the upper (light blue) exciton state
to the lower (dark blue) requires that this specific frequency is available in the spectral density
and that the two excitonic wavefunctions overlap in space. Since the two excitonic wavefunction
is general contain different contributions from local excited states, the relaxation is associated
with a net transfer of energy.
- 175 -
9. Reaction Centers
9.1.The Structure of the Photosynthetic Reaction Center.
1. I ntroduction.
Reaction center complexes convert electronic excitation energy into a stable transmembrane
electrochemical gradient via the process of charge separation. In this section we will discuss their
structure and the mechanism of operation. Reaction centers are integral membrane pigment-
proteins that span the membrane in a vectorial fashion, so that the complex is always oriented the
same way with respect to the sidedness of the membrane. All reaction centers carry out light-
driven electron transfer reactions, resulting in charge separation across the membrane. In
addition, some reaction centers also pump protons. Which are coupled to the electron transfer
reactions involving quinones.
The chemical properties of an excited pigment molecule may be very different from those of the
same molecule in the ground or lowest energy state. In particular the oxidation-reduction
potential for electrons either being added to the molecule or given up by it is very different in the
excited state compared with the ground state (see fig.9.1).
1
When light raises a molecule to an excited electronic state, the
molecule becomes a stronger reductant
LIGHT
I
n
c
r
e
a
s
i
n
g
E
n
e
r
g
y
A* B A B
HOMO
LUMO
LUMO
HOMO
electrons
Fig.9.1. When light raises a molecule to an excited state, the molecule becomes a stronger
reductant.
The net result is that the excited pigment molecule is an extremely strong reducing agent, which
readily gives up an electron to a nearby electron acceptor molecule. This excited state electron
transfer process is the primary photochemical process in photosynthesis and is the moment at
which light energy is converted into chemical energy. In the reaction center of photosynthetic
purple bacteria the product of this light-driven electron transfer event is an oxidized dimer of
chlorophylls (the special pair) and a reduced electron acceptor molecule. Both these species are
ions, with a net charge, and also free radicals with an unpaired electron. The primary ion pair
- 176 -
state consists of the oxidized, and now positively charged, dimer and the reduced and now
negatively charged acceptor. It is, however, a highly unstable system, and the stored energy is
easily lost if it is not stabilized by subsequent secondary processes, discussed below.
The reaction center thus takes light, or electronic excitation energy and uses it to drive electron
transfer reactions. In the instant just after the primary electron transfer process, the system is
poised to at a critical juncture. The oxidized primary electron donor is positioned next to the
reduced acceptor. The most likely outcome according to the laws of thermodynamics is for the
electron to simply transfer back to the donor. This process, called recombination, results in the
conversion of all the energy of the photon into heat, without the opportunity for any of it to be
stored. To avoid this fate, a series of ultrafast secondary electron transfer reactions separate the
oxidized and reduced species in space. The result is that the positive and negative charges
become separated from each other, and the probability of recombination is greatly reduced. If the
secondary stabilization reactions are much faster than the recombination process, then most of
the reaction center complexes will avoid recombination and produce stable products (fig.9.2).
Energetic factors may also be important, as discussed below in section 9.2.
Fig 9.2. (a) Generalized electron transfer scheme in photosynthetic reaction centers. Light
excitation promotes a pigment (P) to an excited state (P*), where it looses an electron to an
acceptor molecule (A) to form an ion-pair state P
+
A
-
. Secondary reactions separate the charges,
by transfer of an electron from an electron donor (D) to P
+
and from the initial acceptor A to the
secondary acceptor (A). (b) Schematic diagram of cyclic electron transfer pathway found in
many anoxygenic photosynthetic bacteria. The vertical arrow signifies light absorption: P
represents the primary electron donor, A, the primary electron acceptor, D and C represent
secondary donors and acceptors. Free energy is conserved by coupling the electron transport to
the transport of protons across the photosynthetic membrane.
2. The Reaction Center Concept.
Now it seems almost self-evident that the reaction center is a well-defined pigment-protein
complex with an identity that is distinct and separable from the rest of the photosynthetic
apparatus, with a fixed composition of protein-subunits, pigments and other cofactors. However,
this view has emerged only slowly during the past 80 years. The idea of the reaction center grew
H
+
-
transport
- 177 -
out of the experiments by Emerson and Arnold in the 1930s which established that many
chlorophylls cooperate to perform the net reactions of photosynthesis. Important early
absorbance difference measurements on purple bacteria by Duysens in the 1950s further laid the
groundwork for the idea that only some of the pigments, that were in a special environment with
special spectroscopic properties were involved in the process of light-driven charge separation.
Later, similar discoveries were made for Photosystem 2 and Photosystem 1 of plants.
The purple bacteria were also the first to have their reaction centers biochemically isolated and
physically characterized. It was the biochemist Clayton who for the first time purified the
reaction center from a carotenoid-less mutant of Rhodobacter sphaeroides (R26). It was the
physicist George Feher who made major contributions to the physical characterization of the R26
reaction center using biochemical, magnetic resonance, laser-spectroscopy and other biophysical
tools.
3. The Purple Bacterial Reaction Center
The purple bacterial reaction centers are located in a specially modified portion of the inner cell
membrane of the organism, called the intracytoplasmic membrane. The membrane invaginates
and sometimes forms tubes, vesicles or flat lamellar membranes, The reaction centers are
oriented uniquely in the membrane, so that the primary electron donor bacteriochlorophylls face
the periplasm space. The electron-donating cytochrome (see below) is then in the periplasmic
space and topologically outside the cell (fig. 9.3).
When these cells are disrupted for study, the broken membranes often reseal into small vesicles
called chromatophores, in the process trapping cytochromes and other periplasmic components
in the interior of the vesicle. Chromatophores are thus opposite in orientation as compared with
intact cells. A detailed artists conception of what such a membrane of a typical photosynthetic
bacterium may look like is shown in fig.9.4.
Fig. 9.3 (a) Cartoon of the photosynthetic membrane of purple bacteria. The photosynthetic
membrane shows many invaginations in which the photosynthetic apparatus is localized.
Topologically, the charge separation occurs from outside to inside. (b) When the cell is
destroyed by pressure or ultrasound, the photosynthetic membrane forms small vesicles,
called chromatophores, with the cytochrome c (C) inside and now charge separation occurs
from inside to outside.
- 178 -
The reaction centers were purified by dissolving the membranes with gentle detergents and
then purifying the pigment-proteins using a variety of biochemical techniques, such as column
chromatography. The purified reaction centers are composed of one copy each of three or four
protein subunits, depending on the species. The constant subunits are known as L (light), M
(medium) and H (heavy). In many species a fourth subunit, known as C (cytochrome) is present.
The designations L, M and H date from the time that the true molecular masses of these subunits
was unknown, the labels being assigned on the basis of the mobility of these peptides as
determined by SDS-PAGE. Ironically, the true masses are significantly different from the
apparent masses, H being the peptide with the lowest mass, L the next and M the heaviest of the
three. However, the L, M and H designations are so well established that it seems best to retain
this nomenclature (for instance the tyrosine on position 210 of the M-subunit is called YM210; a
mutant Tyr->Trp is called YM210W). It also serves as a reminder that SDS-PAGE determined
masses are not very accurate, and that the technique often significantly underestimates masses
for very hydrophobic integral membrane proteins. Table 9.1 gives some compositional data on
reaction center complexes from three representative purple bacteria, Rhodopseudomonas viridis,
Rhodobacter sphaeroides and Chromatium vinosum as well as the filamentous green bacterium
Chloroflexus aurantiacus, which has a similar reaction center.
In addition to the protein subunits, reaction centers contain a number of non-covalently
associated cofactors, including pigments, quinones, metal ions. Purple bacterial reaction centers
contain four molecules of bacteriochlorophyll (BChl), two molecules of bacteriopheophytin
(Bph), one metal ion (in most cases Fe
2+
), two quinones and, in most cases, one carotenoid (see
table 9.1)
Fig. 9.4. Artists view of the photosynthetic membrane of photosynthetic purple bacteria.
Pigment protein complexes (LH2, green; LH1, red; RC, blue) have been inserted in the
invaginations of the photosynthetic membrane. Bottom left: top view of the photosynthetic
membrane: RC (blue) is surrounded by LH1 (red) while LH2(green) is more peripheral.
- 179 -
Table R9.1 Chemical composition of bacterial reaction centers.
Organism Bchl Bph Protein
subunits
Ubiquinone Menaquinone Metal Carotenoid
Rhodopseudomonas
viridis
4 2 4(LMHC) 1 1 Fe 1
Rhodobacter
sphaeroides
4 2 3(LMH) 2 0 Fe 1
Chromatium
vinosum
4 2 4(LMHC) 1 1 Fe 1
Chloroflexus
aurantiacus
3 3 3(LMC) 0 2 Mn 0
High-resolution X-ray structures RCs from two different species have been obtained. The first
was the 4-subunit complex (LMHC) from Rhodopseudomonas viridis. This structure constituted
a landmark in biochemistry, as it was the first time that a high-resolution structure of an integral
membrane protein had been obtained. It earned Michel, Deisenhofer and Huber the 1988 Nobel
prize for chemistry. Subsequently, structures for the 3-subunit (LMH) RC of Rhodobacter
sphaeroides were obtained. The structures for the LMH parts of the two complexes are very
similar. Fig. 9.5 shows this structure for the LMHC complex from Rhodopseudomonas viridis.
1
The reaction center of purple
photosynthetic bacteria has
3 to 4 subunits, depending
on the species
membrane
phospholipid
bilayer
The two central subunits
are integral membrane
proteins with
homologous structures
A small number of
bacteriochlorophylls and
other electron carriers are
bound to the proteins
view parallel to
the membrane
C
L
M
H
- 180 -
Fig 9.5. The structure of the reaction center of Rhodopseudomonas viridis. Apart from the
absence of the Cytochrome (C )subunit, the structure of the reaction center of Rhodobacter
sphaeroides is very similar.
In fig. 9.6, the protein backbone has been removed to show the position of the cofactors in more
detail.
1
The electron carriers in the bacterial reaction center are
arranged around an axis of approximate rotational symmetry
Axis of approximate
rotational symmetry
BChl dimer
BPh
Q
Fe
BChl
BChl
BPh
Q
BChl = bacteriochlorophyll
BPh = bacteriopheophytin
(BChl with 2 H in place of Mg)
Q = ubiquinone
Two of the four BChls
form a dimer (P870)
that acts as the initial
electron donor
Fig 9.6. The arrangement of the cofactors in the bacterial photosynthetic reaction center. Note
the apparent symmetry of the two branches, while light-driven electron transport only occurs
along the A-branch of cofactors.
The L and M subunits of the RC are threaded through the membrane 5 times each, and the
structure of the complex shows that the portion of the protein that crosses the membrane is
almost purely o-helical in secondary structure. The transmembrane helical sections of these
proteins are composed of mostly hydrophobic amino acids. This ensures that the protein is firmly
positioned in the membrane in a unique position and with a well-defined orientation. The H-
subunit is significantly less hydrophobic and has only one transmembrane segment. This anchors
it to the membrane, with the bulk of its mass on the cytoplasmic side. There are thus 11
transmembrane helices, with five each from L and M and one from the H subunit.
The L and M proteins have a pseudo-two-fold axis of symmetry, which is perpendicular to the
plane of the membrane. A pseudo-two-fold axis of symmetry results when the two halves of the
complex are similar but not identical. When the complex is rotated 180 degrees around this
symmetry axis the resulting complex has a structure that is generally similar but not identical to
the original. The symmetry is broken by the H subunit, which has no symmetry-related
counterpart and on a more fundamental level by the fact that the L and M subunits have only 25-
30% sequence homology.
A branch
B branch
- 181 -
The RC protein structure forms a scaffolding upon which the cofactors are arranged. The redox
cofactors are also arranged with a pseudo-two-fold symmetry to form two branches, labeled A
and B in fig 9.6. A variety of evidence indicates that light-driven electron transfer proceeds only
down the pigments on the A branch and then crosses over from the A to the B branch at the level
of the quinones. The function of the pigments in the B-branch is not clear and may be primarily
structural.
The two pigments labeled D
A
and D
B
in fig. 9.6 are BChl a molecules that are very closely
interacting. They are called P870, after the wavelength maximum of their Q
y
absorbance band.
This is 870 nm for those purple bacteria that contain BChl a and 960 nm for those that contain
BChl b. This pair of BChls was identified as a special pair and together they form the
photoactive pigment whose excited state looses an electron to form the primary charge
separated state. Once the charge separation has occurred P870
+
is formed, a state in which the
photoactive pigment has lost one electron. This oxidized state, with one unpaired electron
remaining, has been studied extensively with magnetic resonance techniques (ESR, electron spin
resonance, ENDOR, electron nuclear double resonance), which are sensitive to the properties of
the unpaired electron that remains on the special pair after the electron transfer forms the cation
radical. The remaining unpaired electron is approximately equally shared between the two
pigments, resulting in a reduction of the electron-nuclear hyperfine coupling constants by a
factor of 2, compared with the cation radical of oxidized monomeric BChl a in solution.
The C (cytochrome) unit is found in many purple bacterial RCs, but it is absent in others. It is
not, strictly speaking, an integral membrane protein, as it has no transmembrane segments. Its
entire mass is found on the periplasmic side of the membrane. However, in Rhodopseudomonas
viridis RCs the C-subunit is tightly associated with the rest of the complex. Also, in some cases,
a covalently attached fatty acid molecule serves to anchor the C-subunit to the membrane. The
most distinctive feature of the C-subunit is the presence of the four heme groups, which are
covalently attached to cysteine amino acid side chains. The hemes alternate with high and low
redox potentials, with the highest-potential heme near the LM-complex and the lowest-potential
heme at the end of the chain. Soluble cytochrome c
2
donates an electron to the low-potential
heme at the end of the chain and the electron is subsequently transferred through the other hemes
until it reduces the oxidized special pair.
The kinetics and energetics of this process are remarkable. The midpoint redox potential of the
electron donor cytochrome c
2
is +285 mV, while the first acceptor has an potential of -60mV.
This particular electron transfer step is thus energetically uphill by +345 mV, yet it still takes
place in 60 ms when the cytochrome c
2
is bound to the tetraheme cytochrome subunit, because of
a favorable positioning of the two heme groups involved. In those species of purple bacteria that
lack a bound C-subunit, the soluble cytochrome c
2
donates electrons directly to the oxidized
special pair, after first docking to the periplasmic surface of the RC.
- 182 -
9.2. Primary Electron Transfer Reactions in Bacterial Photosynthetic
Reaction Centers
A wide variety of techniques has been used to investigate the bacterial RC system, including
almost every kind of spectroscopy, as well as a range of biochemical and genetic manipulations.
Here it is only possible to give a brief summary of the most important results. The focus will be
on the first few reactions that follow the excitation of the special pair, either directly by photon
absorption or indirectly by excitation energy transfer from the antenna or other reaction center
pigments. The technique of ultrafast transient absorption spectroscopy (see fig 9.7) has been
especially informative with respect to elucidating the pathway of electron flow in these RCs and
will therefore be emphasized.
1
The sequence and kinetics of the initial electron-transfer reactions
can be studied by exciting RCs with short pulses of light
sample
excitation
pulses
probe
pulses
measure the intensity
of the transmitted
probe beam averaged
over many pulses
vary the path
length for the
probe pulses
A
b
s
o
r
b
a
n
c
e
c
h
a
n
g
e
Delay between excitation
and probe pulses
1
2
Laser
beam
splitter
1
and
2
: different
probe wavelengths
detect different
electron carriers.
train of short pulses
Fig 9.7. Layout of a femtosecond transient absorption experiment. Note that the time delay is
simply (and precisely) determined by the difference in path length between pump and probe
pulses and controlled by the position of a retroreflector.
Fig.9.8 summarizes the kinetics and energetics of the primary photochemistry and early
secondary reactions that take place in isolated RCs. A variety of evidence indicates that the
electron transfer pathways and kinetics in isolated RCs are not significantly altered from their
behavior in vivo.
- 183 -
1
BChl dimer
BPh
Q
Fe
BChl
3 ps
1 ps
200 ps
LIGHT
When the complex is excited with light, an electron moves
from the BChl dimer (P870) to a BPh and then to a quinone
A branch
B branch
Fig 9.8. Sequence of electron transfer steps along the active branch of cofactors in the bacterial
reaction center
Following excitation of the special pair, the excited state of P870 (P960 in organisms that contain
BChl b, such as Rps. viridis) has a lifetime of about 3 ps at room temperature, decreasing to
about 1 ps at cryogenic temperatures. This excited state (P870*) decay is most conveniently
monitored by measuring stimulated emission in the 900 nm region. The P870* excited state is a
very strong reductant, with an estimated excited state redox potential of -940 mV vs. NHE. It
decays primarily by loosing an electron to become the cation radical P870
+
. Whenever a pigment
is excited or either gains or looses an electron, its ground state absorption bands will bleach. New
absorption bands that are characteristic of the excited, oxidized or reduced species will appear at
the same time as the ground state bands bleach. Analysis of these spectral changes helps
elucidate the electron transfer pathway.
As P870* decays to form P870
+
, absorbance bands bleach at 535 nm and 760 nm, which are
assigned to the Q
x
and Q
y
transitions of the bacteriopheophytin on the A branch. At the same
time a broad new absorbance band appears in the 650 nm region, which is assigned to the
formation of the anion radical BPh
A
-
. The new state formed is the radical ion-pair P870
+
BPh
A
-
, in
which an electron has been transferred to the bacteriopheophytin on the A branch. The A branch
bacteriopheophytin, instead of the one of the B branch, is identified as the acceptor largely by the
difference spectra in the Q
x
spectral region. The two bacteriopheophytins have slightly different
spectral signatures, due to differences in the amino acid polarities in their immediate
surroundings. If it were not for this fortuitous spectral difference between the two BPh
molecules, it would be very difficult to determine that one electron transfer branch is active
while the other is inactive.
- 184 -
The P870
+
BPh
A
-
ion-pair state decays in 200-300 ps to form the state P870
+
Q
A
-
. Finally the
electron is transferred to Q
B
in 100-200 s to form the state P870
+
Q
B
-
. This state is characterized
by bleaching of the absorption bands due to P870 and Q
B
, plus the appearance of new bands due
to the cation and the anion, respectively. The Q
A
-
Q
B
-
reaction is thermally activated, while all
the others are nearly independent of temperature, in fact speeding up slightly at cryogenic
temperatures. A variety of inhibitors, including herbicides such as atrazine, block this electron
transfer process by displacing Q
B
from the binding site. Proton uptake accompanies electron
transfer in the quinone complex.
In parallel with the Q
A
-
Q
B
-
reaction, electron transfer from a cytochrome rereduces the
oxidized special pair. This is either the bound cytochrome subunit (C) in those organisms that
contain it, or a small soluble cytochrome, in most cases cytochrome c
2
. The bound cytochrome
donates an electron in a reaction that takes only a few hundred ns and which also continues to
take place at cryogenic temperatures. At low (<120K) the rate of this reaction becomes T-
independent (about 1 ms) , and this T-independence is clear evidence for a tunneling process,
which was later identified as nuclear tunneling (see below).
The arrangement of electron transfer cofactors as shown in fig 9.6 suggests that an earlier, charge
separated state may precede the formation of the P870
+
BPh
A
-
state. This earlier state
P870
+
BChl
A
-
, where BChl
A
is the accessory bacteriochlorophyll labeled B
A
, and is likely
because of the spatial position of BChl
A
between P870 and BPh
A
. This state almost certainly
participates in the primary electron transfer process, although the nature of its involvement has
been a matter of intense debate. If such a state is formed, then the data clearly indicate that the
rate constant for its decay is faster than that of its formation, thereby precluding the buildup of
substantial (measurable) amounts of the state. The energy of the P870
+
BChl
A
-
state is not
known very well. The most important question is whether it lies above or below the P870*
state. If it lies above the P870* state, then it probably contributes to the primary charge
separation by what is known as a superexchange mechanism, in which the BChl
A
anion can be
viewed as virtual intermediate that mixes with the P870* state and thus provides a path for
charge separation to form P870
+
BPh
A
-
. Most evidence indicates, however, that the role of the
BChl
A
is that of a real electron carrier, that is populated to a small degree because of the fast
(<1 ps) electron transfer from BChl
A
-
to form BPh
A
-
. The distances and calculated couplings
between the special pair and BPh
A
are sufficiently long and weak, resp., that it is extremely
unlikely that the electron transfer process takes place directly from P870* to BPh
A
(see below).
Perhaps the most striking aspect of the comparison of the structural and kinetic data on RCs is
the apparent two-fold symmetry of the structure, contrasted with the clear evidence for strong
asymmetry in the electron transfer pathway. Estimates of the ratio for electron transfer
probability down the A branch compared with the B branch are about 100:1. A considerable
effort, including analysis of many engineered mutants, has gone into trying to understand the
factors that direct electron transfer down the A branch instead of the B branch. Although a final
answer is not (yet) there, it seems clear that the tuning of free energy of the states P870
+
BChl
A
-
and P870
+
BPh
B
-
by the protein environment plays a crucial role. On top of this, also the
functional role of this unidirectional electron transfer is not clear, and it may easily be that, for
instance in Photosystem 1 of green plants light-driven charge separation occurs along both
branches of cofactors.
- 185 -
Another and important aspect of the kinetic constants shown in fig 9.8 is that the rates of the
recombination processes, in which the electron returns from one of the acceptors directly to the
oxidized special pair, are invariably a factor of 50 or more slower than are the rate constants for
the forward reaction. It is because of this kinetic control of the rates of wasteful processes that
the quantum yield for photochemistry is so high. This kinetic steering of the system towards the
productive sequence of charge-separation reactions and away from the wasteful recombination
reactions is the critical factor that makes the primary processes of photosynthesis so efficient.
Again, the molecular reasons for this remarkable behavior have proven difficult to elucidate. One
of the major factors thought to be involved in the slow recombination reactions is the large
energy gap involved. This is best understood by considering some theoretical aspects of
biological electron transfer.
- 186 -
9.3. Biological Electron Transfer: The Marcus model
I ntroduction
Biological electron transfer reactions have been the subject of a great deal of both experimental
and theoretical analysis in a wide range of systems. Of these systems the bacterial photosynthetic
reaction center (RC) is attractive because it exhibits in a single complex a number of unusual
features, including the ability to follow the electron transfer over 13 orders of magnitude in time
(sub ps-seconds), to vary the temperature from liquid helium to above room temperature, and to
modify the free energies involved in the process by chemical, genetic and electrical methods.
The structure of the RC (and many mutants) is known at high resolution, diffusion processes are
not involved, and a wealth of kinetic and other spectroscopic data is available. No other
biological system possesses all these features.
The Marcus equation
The theoretical description of biological electron transfer has grown out of the pioneering work
of Marcus on inorganic reactions in solution, for which he received the 1992 Nobel prize for
chemistry. The electron transfer process can be viewed as a nonradiative relaxation process from
an initial state with the electron donor (D) reduced and the acceptor (A) oxidized to a final state
with the donor oxidized and the acceptor reduced, according to Eq.9.1.
red ox ox red
A D A D + +
(9.1)
The Fermi Golden rule describes the first-order rate constant for the electron transfer process,
according to Eq. 9.2
( )
f i f i et
E E V k = o
t
2
2
(9.2)
This equation is in many ways similar to that for the Frster theory for energy transfer discussed
above. An initial state in which an electron (or an excitation) is localized makes a nonradiative
transition to a final state in which the electron (or excitation) has moved to another molecule. In
Eq. 9.2, the transition is between two particular microscopic states of the system, which includes
details such as the vibrational state and the electronic properties. Note that the wavefunctions for
the initial and final states (
i
and
f
) include both the donor and the acceptor. The parameter V
is the electronic coupling between the two states and has units of energy, usually given in wave
numbers (cm
-1
). The delta function ensures conservation of energy. This means that only initial
- 187 -
and final states of the same energy contribute to the observed rate (see fig.9.9).
Nuclear Dynamics and Electron Transfer
DA
D
+
A
-
Nuclear Configuration
FC V W
RP
2 2
t
=
Rate of electron Transfer:
R
DA
R
D
+
A
-
R
#
Fig 9.9. Electron transfer from a donor to an acceptor.
In Fig 9.9 the initial state is indicated as DA, the final state as D
+
A
-
. In DA the electron is in a
bound state on D, in D
+
A
-
it is in a bound state on A. The free energy dependence of DA and
D
+
A
-
on the nuclear configuration is given by the parabolic curves. Note how the electron
transfer is coupled to the motion of the nuclei. When the system is at R
DA
, the energetic
minimum of DA, the electron transfer will never take place, because for that nuclear
configuration the energy of D
+
A
-
is much too high (see fig 9.10). To cross efficiently from DA to
D
+
A
-
the system must first move to R
#
(symbolized in fig 9.9 by the wavy green line) where DA
and D
+
A
-
have equal energy . To do so the system must be in some higher vibrational state
(activated), and only then is there a significant probability that the system will cross from DA
to D
+
A
-
. Moreover, since the nuclei are stationary on the timescale of electron transfer the
electron transfer can only take place when the initial and final state have the same nuclear
configuration. The probability that this is the case is measured by the overlap between the initial
and final state vibrational wavefunctions (like in the Franck-Condon principle).
- 188 -
Correspondence between the
electronic energy levels (shown on
the left) and the nuclear energy
levels (shown on the right).
(a) At the nuclear configuration q
0
R
the
electron to be transferred in DA is in an
occupied electronic energy level (denoted by
the blue circle) and the lowest unoccupied of
D
+
A
-
(denoted by an unfilled circle) is too
high in energy to be a good electron acceptor.
(b) As the nuclei rearrange to a configuration
represented by q
*
, DA and D
+
A
-
become
degenerate and electron transfer occurs by
electron tunneling through a barrier of height
V and width r, the edge-to-edge distance
between D and A. (c) The system relaxes to
the equilibrium position of D
+
A
-
denoted by
q
0
P
in which the lowest unoccupied electronic
level of DA is higher in energy than the
highest occupied electronic level of D
+
A
-
R
q
0
Fig 9.10. Correspondence between electronic energy levels (left) and potential energy curves
(right). Note that only at the crossing point the electron can be transferred from D to A.
However, there are many possible initial states and many possible final states of the system.
Many of those will only differ by the vibrational states of the molecules. In order to recover the
overall rate of electron transfer, it is necessary to sum over all possible initial states and also to
weigh their contributions according to how likely they are to contribute. This is given in Eq.9.3.
( )
f i f i
i
i et
E E V P k =
o
2
(9.3)
In Eq.9.3 the summation is over all the vibrational substates of the initial state i, weighted
according to their probability
i
P times the square of the electron transfer matrix element. Making
the Born-Oppenheimer approximation for the separation of nuclear and electronic wavefunctions
(nuclear motion is much slower than electronic motion) simplifies this equation. This results in
Eq.9.4
FC V k
el et
2
2
t
=
(9.4)
in which
2
el
V is the square of the electronic coupling matrix element between the electronic states
of the reactant and those of the product, and FC is the Franck-Condon factor (implicitly
including the summation over all states).
- 189 -
The electronic coupling matrix element experimentally depends primarily on the distance and
orientation of the reacting species. A variety of evidence indicates that this parameter depends
exponentially on the distance between the reacting groups, as described by Eq.9.5
) exp(
2
0 ,
2
d V V
el el
| =
(9.5)
In Eq.9.5
2
0 , el
V is the maximum possible electronic coupling when the molecules are in contact,
| is a parameter describing the distance dependence of the coupling, and d is the distance. There
has been a great deal of discussion about the proper value of | , including whether it is even
appropriate to have a single parameter that describes the distance dependence. A consensus
seems to have been reached that a | value of 1.4
-1
is reasonable for protein-mediated
electron transfer processes. Note the difference between the exponential distance dependence of
electron transfer and the inverse sixth power dependence of the Forster energy transfer process.
This difference arises because the Frster process is mediated by a dipole-dipole coupling
interaction, whereas the electron transfer is mediated by coupling that depends on the spatial
overlap of the wavefunctions (the electron must be transferred from the donor to the acceptor!!)
of the reacting species, which characteristically fall off in an exponential manner.
The Franck-Condon factor depends on the overlap of the nuclear wavefunctions of the initial and
final states, suitably weighted by the Boltzmann factor
i
P . Again, this term is almost identical to
the Franck-Condon factor discussed in the context of electronic transitions. This term includes
the effects of temperature and the free energy change of the reaction. Unfortunately this term is
difficult to calculate, although simplifications are available. A useful way to describe the Franck-
Condon factor is by means of the reorganization energy which can be considered as the
amount of energy required to distort the geometry of the reactants into that of the products
without the electron transfer actually taking place (see fig.9.11). A simple equation describing
how the Franck-Condon factor depends on the energy change in the reaction was proposed by
Marcus (based on parabolic free energy surfaces for reactants and products as a function of the
reaction coordinate, see fig.9.11), and is given in Eq.9.6)
( ) ( ) | | kT G kT FC t 4 exp 4
2
0 2 1
A =
(9.6)
where
0
G A is the standard free-energy change in the reaction (NB E G A = A
0
).
- 190 -
Calculation of the Activation Energy AG
#
Reorganization energy :
Energy required to move from equilibrium nuclear
reactant to equilibrium nuclear product configuration
without electron transfer.
Calculating the
thermal Activation
energy:
4
) (
2
#
E
G
A
= A
Fig.9.11. The energies of the initial (DA) and final state (D
+
A
-
) are represented by the parabolic
curves V
R
and V
P
. The reorganization energy is the amount of energy that one must add to the
initial state to bring it to the configuration of the final state. It is simple to calculate the classical
activation energy AG
#
as a function of the reorganization energy and the free energy
difference AE.
Three general regimes can be considered for the free-energy dependence of the electron transfer
rate constant, shown schematically in fig.9.12
- 191 -
AG < AG =
AG >
Marcus parabola
Activated Activationless Inverted
Fig 9.12. Three regimes of the free energy dependence of the electron transfer rate. A. For
AG< the electron transfer is activated and will slow down as a function of temperature. For
AG= the potential energy curve of D
+
A
-
crosses that of DA in the bottom of the parabola.
There is almost no T-dependence, in fact the rate will speed up upon lowering T because of the
increased probability to find the system in the lowest vibrational level. For AG>, again a
barrier has to be overcome and as a consequence the rate slows down and becomes thermally
activated. The Marcus model for electron transfer results in a parabolic dependence of the rate
of electron transfer on the free energy difference of the reaction (see the frame labeled Marcus
parabola) and this relation has been checked extensively in the bacterial RC. Finally it is not
difficult to see that in case AG>, nuclear tunneling contributions may significantly contribute to
the electron transfer rate making the parabola less steep on the high AG side(dashed curve)
If < A
0
G the process will exhibit normal thermally activated behavior, illustrated by
fig.9.12A. This is the case for endergonic or slightly exergonic reactions. If the reaction is
significantly exergonic, so that = A
0
G , then the potential energy curves for the reactants and the
products intersect near the bottom of the reactant potential well, and the reaction is in the
activationless regime (fig.9.12B). The Franck-Condon factor is at a maximum and the reaction
is very fast with a very weak temperature dependence. Remarkably, the earliest reactions in
photosynthetic RCs are all in this regime. Finally, for extremely exergonic reactions with
> A
0
G , the system is in the inverted regime, and the Franck-Condon factor, and therefore
the rate, decreases again (fig.9.12C).
The size of the Marcus parameter depends on the nature of the reacting groups and their local
environment (polar, non-polar, hydrophobic, hydrophilic, H-bonds, water etc). If both the donor
and the acceptor are large molecules such as chlorophylls, which do not change their structure or
- 192 -
vibrational frequencies much upon oxidation or reduction and if they in a similar environment,
then the value for will be relatively small, typically a few tenths of an eV. If, however, the
molecules are very different, such as one being a quinone and the other being a chlorophyll and
both are in a very different environment, then the value for can be very large, as much as 1.5
eV.
Nuclear Tunneling and Temperature Dependence
When the temperature is sufficiently high to perform the electron transfer systems will be
activated to the crossing point R#. Note that when the system reaches R# the electron will tunnel
from a bound state on D to a bound state on A. On the other hand, when the temperature is
lowered the system can no longer cross the barrier
#
G A and the electron transfer reaction slows
down. However, since even in the lower vibrational states the system has a finite chance to be
outside the parabolic potential energy well, there is a probability that the system will tunnel into
the barrier, R# will be reached and electron transfer may occur. This phenomenon is called
nuclear tunneling, in which the system tunnels from the configuration DA to the configuration
D
+
A
-
. During this slow nuclear tunneling event the electron will tunnel from the donor to the
acceptor, very similar to what happened in the activated case.
Nuclear Tunneling and theQuantumMechanical ET Rate
Activated
NuclearTunneling
( ) ( ) ( )
|
.
|
\
|
=
=
e
e e _ _
t
1
exp 1 exp
2
2
0
'
2
T k T k n V W
B B
n
n n RP
Franck-Condon
factors
Boltzmann Distribution
of Oscillators
Density of
states
n
_ ' n
_
FC V W
RP
2 2
t
=
Rate of electron Transfer:
Fig 9.13. Illustration of nuclear tunnelling. Instead of crossing the barrier in point C
(R
#
), for which the system must be activated, it may tunnel through the barrier from
the R (DA) to the P (D
+
A
-
) state. The tunnelling event now implies a change in the
configuration of the nuclei and is called nuclear tunnelling. The expression for W is
used to calculate the rate of electron transfer quantum mechanically, including the
tunnelling contribution.
- 193 -
Electron transfer in the bacterial reaction center
The bacterial reaction center displays electron transfer reactions over a time range of
picoseconds to tens of seconds (10
-12
s-10 sec). The driving forces for these reactions could be
varied by (1) mutagenesis in which certain amino acids in the neighborhood of the cofactors
were modified, thereby changing the oxidation/reduction potential of that cofactor and (2)
chemical substitution. The latter was extensively done for the quinones Q
A
and Q
B
. Thus electron
transfer rate constants were obtained for a number of values of the driving force and the resulting
free energy dependence was modeled using the QM-version of the Marcus expression (see fig
9.14). Note how well the model reproduces the data. Note also that in a number of case electron
transfer occurred in the Marcus inverted region.
Picoseconds (10
-12
sec)
10 seconds
Free energy dependence of electron transfer
Fig 9.14. Free energy dependence of all electron transfer in the bacterial reaction centre
You might also like
- Biological Inorganic Chemistry - Structure and Reactivity - Ivano Bertini Et Al. (University Science Books, 2007)Document597 pagesBiological Inorganic Chemistry - Structure and Reactivity - Ivano Bertini Et Al. (University Science Books, 2007)Julio Cesar Costa100% (3)
- Staelin, David H - Electromagnetics and Applications-Massachusetts Institute of Technology (2011)Document443 pagesStaelin, David H - Electromagnetics and Applications-Massachusetts Institute of Technology (2011)Marlon Recarte100% (2)
- Understanding Creation From The QuranDocument168 pagesUnderstanding Creation From The QuranZaid Ghazzawi100% (6)
- 11 Photosynthesis-SDocument14 pages11 Photosynthesis-Sqwerty100% (1)
- Gut CP 2018 Ed Book WebDocument1,843 pagesGut CP 2018 Ed Book WebjoseNo ratings yet
- Advances in Optics Vol 5Document650 pagesAdvances in Optics Vol 5Tayyab ImranNo ratings yet
- Determination of The Electronic and Structural Configuration of Coordination Compounds by Synchrotron-Radiation TechniquesDocument57 pagesDetermination of The Electronic and Structural Configuration of Coordination Compounds by Synchrotron-Radiation TechniquesIgor GomesNo ratings yet
- T2 Skript BertlmannDocument186 pagesT2 Skript BertlmannFaulwienixNo ratings yet
- 00012Document1 page00012mazharNo ratings yet
- Quantum Computing With NMR (Progress in Nuclear Magnetic Resonance Spectroscopy, Vol. 59, Issue 2) (2011)Document30 pagesQuantum Computing With NMR (Progress in Nuclear Magnetic Resonance Spectroscopy, Vol. 59, Issue 2) (2011)sepot24093No ratings yet
- free-Chemistry-Notes Gulag FreeDocument90 pagesfree-Chemistry-Notes Gulag FreeNameanxa AngelsNo ratings yet
- Fabrizio M A Course in Quantum Manybody TheoryDocument350 pagesFabrizio M A Course in Quantum Manybody TheoryStrahinja DonicNo ratings yet
- Advanced Synthesis of Gold and Zirconia Nanoparticles and their CharacterizationFrom EverandAdvanced Synthesis of Gold and Zirconia Nanoparticles and their CharacterizationNo ratings yet
- 1 s2.0 S0079670010001139 MainDocument113 pages1 s2.0 S0079670010001139 Mainไตเติ้ล สบม.No ratings yet
- Nikolai Vekshin - Biophysics of Mitochondria-Springer International Publishing - Springer (2019)Document205 pagesNikolai Vekshin - Biophysics of Mitochondria-Springer International Publishing - Springer (2019)Leandro Dellacqua100% (1)
- 3 Mod 3 Nuclear ParametersDocument66 pages3 Mod 3 Nuclear ParametersSaad Javaid SiddiqueNo ratings yet
- Retention and Selectivity in Liquid Chromatography: Prediction, Standardisation and Phase ComparisonsFrom EverandRetention and Selectivity in Liquid Chromatography: Prediction, Standardisation and Phase ComparisonsNo ratings yet
- PhDThesis Scheuring ScreenDocument115 pagesPhDThesis Scheuring Screenodiaz01100% (1)
- Physics Notes2016v15 PDFDocument291 pagesPhysics Notes2016v15 PDFCurtis Patterson100% (1)
- Roy Choudhury, D - Networks and Systems-New Academic Science Limited (2014)Document959 pagesRoy Choudhury, D - Networks and Systems-New Academic Science Limited (2014)Prakash VermaNo ratings yet
- DOCDocument1 pageDOCDennoh OlengoNo ratings yet
- Nuclear Physics and Reactor Theory Vol 1 and 2Document245 pagesNuclear Physics and Reactor Theory Vol 1 and 2kollliNo ratings yet
- Atomic and Nuclear PhysicsDocument254 pagesAtomic and Nuclear PhysicsAli AhmedNo ratings yet
- ICP-OES Operation Training CourseDocument74 pagesICP-OES Operation Training CourseFrankNo ratings yet
- ST Nar 19Document50 pagesST Nar 19popa.irinaNo ratings yet
- Equations in PhysicsDocument105 pagesEquations in PhysicsLian ChiNo ratings yet
- t1Document4 pagest1romulo_moraes_silvaNo ratings yet
- IARE - SM - Lecture NotesDocument86 pagesIARE - SM - Lecture NotesPrajwal B NaikNo ratings yet
- Notes On General Chemistry, 2e: Chapter 1, The Stuff of ChemistryDocument12 pagesNotes On General Chemistry, 2e: Chapter 1, The Stuff of ChemistryKeturah aNo ratings yet
- Full Download Book Ideas of Quantum Chemistry Volume 1 From Quantum Physics To Chemistry PDFDocument41 pagesFull Download Book Ideas of Quantum Chemistry Volume 1 From Quantum Physics To Chemistry PDFtommy.peacock526100% (16)
- Orgo SolutionsDocument151 pagesOrgo SolutionsBarryNo ratings yet
- John Dirk Walecka: World ScientificDocument11 pagesJohn Dirk Walecka: World ScientificArshad PathanNo ratings yet
- Applied I ModuleDocument220 pagesApplied I ModuleshoyaNo ratings yet
- Gelombang Dan Optik S2 PDFDocument188 pagesGelombang Dan Optik S2 PDFEvy Syafitri HarahapNo ratings yet
- Quantum Dynamics Applications in Biological and Materials Systems (Eric R - Bittner)Document336 pagesQuantum Dynamics Applications in Biological and Materials Systems (Eric R - Bittner)Phyo ZawNo ratings yet
- Analogue Electronics 1, DEE 1 Notes - 075733Document137 pagesAnalogue Electronics 1, DEE 1 Notes - 075733Kirimi DanNo ratings yet
- Tfy-3.365 Statistical Physics and Thermodynamics (Spring 2004)Document3 pagesTfy-3.365 Statistical Physics and Thermodynamics (Spring 2004)Umer IlyasNo ratings yet
- Classical ElectrodynamicsDocument592 pagesClassical ElectrodynamicsŞenol TopuzNo ratings yet
- Physical - Anal1 2022Document59 pagesPhysical - Anal1 2022John Yaw AmankrahNo ratings yet
- LIGHTING BASICS Practical Application Gu 2Document214 pagesLIGHTING BASICS Practical Application Gu 2muktharNo ratings yet
- Mechanochemistry in Nanoscience and Minerals EngineeringDocument422 pagesMechanochemistry in Nanoscience and Minerals EngineeringRomán Hermosillo Mendoza100% (1)
- SMCDocument326 pagesSMCEdgardo Olmedo Sotomayor0% (1)
- Ergodic NotesDocument115 pagesErgodic NotesoveiskntuNo ratings yet
- Theoretical Statistical Physics: Prof. Dr. Christof Wetterich Institute For Theoretical Physics Heidelberg UniversityDocument175 pagesTheoretical Statistical Physics: Prof. Dr. Christof Wetterich Institute For Theoretical Physics Heidelberg UniversityankurNo ratings yet
- Mechanochemistry in Nanoscience and Minerals EngineeringDocument422 pagesMechanochemistry in Nanoscience and Minerals EngineeringJazmin RuizNo ratings yet
- Shen S.-Q. Lecture Notes On Quantum Mechanics (Hong Kong, Web Draft, 2004) (265s) - PQMDocument265 pagesShen S.-Q. Lecture Notes On Quantum Mechanics (Hong Kong, Web Draft, 2004) (265s) - PQMfajnmanNo ratings yet
- Finalized Module For Classical Mech IIDocument157 pagesFinalized Module For Classical Mech IIchilotawNo ratings yet
- Modern Optical SpectrosDocument510 pagesModern Optical SpectrosAlberto Jaimes PitaNo ratings yet
- Polymer - Chemistry - (Contents)Document8 pagesPolymer - Chemistry - (Contents)金衍扜No ratings yet
- Full Download Book Dynamics of Molecular Excitons Theories and Applications Nanophotonics PDFDocument41 pagesFull Download Book Dynamics of Molecular Excitons Theories and Applications Nanophotonics PDFjuan.starkey551100% (14)
- Tumulka R Foundations of Quantum MechanicsDocument478 pagesTumulka R Foundations of Quantum MechanicsStrahinja DonicNo ratings yet
- Full TextDocument199 pagesFull Textbayou71usaNo ratings yet
- Phy 2511 Classical Mechanics Module Edited 18-04-2017 PDFDocument110 pagesPhy 2511 Classical Mechanics Module Edited 18-04-2017 PDFlitebele litebeleNo ratings yet
- Ligand Coupling Reactions with Heteroatomic CompoundsFrom EverandLigand Coupling Reactions with Heteroatomic CompoundsRating: 4 out of 5 stars4/5 (1)
- Temperley-Lieb Recoupling Theory and Invariants of 3-Manifolds (AM-134), Volume 134From EverandTemperley-Lieb Recoupling Theory and Invariants of 3-Manifolds (AM-134), Volume 134No ratings yet
- Photosynthesis Under Stress 2013Document28 pagesPhotosynthesis Under Stress 2013naufal samiNo ratings yet
- 5 FotosintesiDocument29 pages5 FotosintesiSerena DamianNo ratings yet
- Ecophysiology of Papaya: A Review: Eliemar Campostrini and David M. GlennDocument12 pagesEcophysiology of Papaya: A Review: Eliemar Campostrini and David M. GlennKIRAN V RNo ratings yet
- General Biology 1: First Semester-Quarter 2Document21 pagesGeneral Biology 1: First Semester-Quarter 2Laelannie MagpayoNo ratings yet
- Tredici Photobiology 2010Document20 pagesTredici Photobiology 2010Luis EspinozaNo ratings yet
- Cell BioenergeticsDocument42 pagesCell BioenergeticsSh SarkerNo ratings yet
- Biology Unit 2 For Cape ExaminationsDocument11 pagesBiology Unit 2 For Cape ExaminationsDexan LewisNo ratings yet
- FTIR Spectroscopy PhotRes2009Document15 pagesFTIR Spectroscopy PhotRes2009DickyNo ratings yet
- Photosynthesis in Higher Plants - by @MadXAbhiOfficialDocument11 pagesPhotosynthesis in Higher Plants - by @MadXAbhiOfficialmintukumar696969No ratings yet
- PHOTOSYNTHESISDocument17 pagesPHOTOSYNTHESISDave Degaetano100% (3)
- Eddie M. Raguindin: Biology TeacherDocument23 pagesEddie M. Raguindin: Biology TeacherMhimi ViduyaNo ratings yet
- TMP 8 FD1Document38 pagesTMP 8 FD1FrontiersNo ratings yet
- Photosynthetic Reaction Centers - So Little Time, So Much To DoDocument31 pagesPhotosynthetic Reaction Centers - So Little Time, So Much To DoGyamfi Atta KwameNo ratings yet
- Jurnal Organologam PDFDocument31 pagesJurnal Organologam PDFdyahyektiindrajatiNo ratings yet
- Forum: Preface On Making OxygenDocument3 pagesForum: Preface On Making OxygenMuhamad Ivan AbrorNo ratings yet
- Ch10 Test File-PhotosynthesisDocument40 pagesCh10 Test File-PhotosynthesisAnonymous 9xheGVRM1No ratings yet
- Cell and Molecular Biology Concepts and Experiments 7th Edition Karp Test Bank DownloadDocument37 pagesCell and Molecular Biology Concepts and Experiments 7th Edition Karp Test Bank DownloadBen Flott100% (21)
- How Does Chloroplast Protect Chlorophyll Against EDocument18 pagesHow Does Chloroplast Protect Chlorophyll Against EIrene MartinezNo ratings yet
- 12 Rna World 2nd PDFDocument34 pages12 Rna World 2nd PDFedeceNo ratings yet
- (Lecture Notes in Chemistry 92) Giacomo Bergamini, Serena Silvi (Eds.) - Applied Photochemistry - When Light Meets Molecules-Springer International Publishing (2016)Document536 pages(Lecture Notes in Chemistry 92) Giacomo Bergamini, Serena Silvi (Eds.) - Applied Photochemistry - When Light Meets Molecules-Springer International Publishing (2016)yulliarperezNo ratings yet
- Essentials of The Living World 5th Edition George Johnson Solutions Manual 1Document4 pagesEssentials of The Living World 5th Edition George Johnson Solutions Manual 1ronald100% (37)
- 044BIO11-13 Photosynthesis in Higher Plants (Study Notes)Document12 pages044BIO11-13 Photosynthesis in Higher Plants (Study Notes)Zen FredyNo ratings yet
- Plant Nanobionics A Novel Approach To Overcome The Environmental ChallengesDocument11 pagesPlant Nanobionics A Novel Approach To Overcome The Environmental ChallengesCuriousNo ratings yet
- George S. Bullerjahn & Anton F. Post Eds. - Physiology and Molecular Biology of Aquatic CyanobacteriaDocument128 pagesGeorge S. Bullerjahn & Anton F. Post Eds. - Physiology and Molecular Biology of Aquatic CyanobacteriaGrasic100% (1)
- Photochemical Applications of Solar Energy - Photocatalysis and Photodecomposition of WaterDocument85 pagesPhotochemical Applications of Solar Energy - Photocatalysis and Photodecomposition of Watereimaiokanenas100% (1)
- Unit 6 Notes Csir NetDocument163 pagesUnit 6 Notes Csir NetAlma VNo ratings yet
- CHAPTER 21 PhotosynthesisDocument12 pagesCHAPTER 21 Photosynthesis楊畯凱No ratings yet
- Photosynthesis - WikipediaDocument28 pagesPhotosynthesis - WikipediakamaalNo ratings yet