You might also like
- Mass Spectroscopy and Raman Spectroscopy.Document27 pagesMass Spectroscopy and Raman Spectroscopy.kritika100% (4)
- UV-VIs SpectrosDocument134 pagesUV-VIs SpectrosSaumya Prasad100% (1)
- Quantitative Human Physiology: An IntroductionFrom EverandQuantitative Human Physiology: An IntroductionRating: 2 out of 5 stars2/5 (1)
- Unit 7 Flame PhotometryDocument32 pagesUnit 7 Flame PhotometryRia Agnez100% (8)
- NMR Spectroscopy - Short NoteDocument6 pagesNMR Spectroscopy - Short Notecoolhemakumar100% (4)
- SpectrosDocument28 pagesSpectrosPraveen Kumar AvvaruNo ratings yet
- Instrument Control Uning LABVIEWDocument69 pagesInstrument Control Uning LABVIEWcoolhemakumarNo ratings yet
- Chem Final Notes UsydDocument5 pagesChem Final Notes UsydRobs50% (2)
- Analytical Chemistry Lecture Pre-Limenaries MT 2ADocument10 pagesAnalytical Chemistry Lecture Pre-Limenaries MT 2AJerica Mae GabitoNo ratings yet
- Mass SpectrometryDocument4 pagesMass SpectrometryCarlton GrantNo ratings yet
- Analytical Chemistry Quiz MaterialDocument11 pagesAnalytical Chemistry Quiz MaterialAltaf Ur RehmanNo ratings yet
- Analytical MethodsDocument16 pagesAnalytical MethodsSubhash Dhungel100% (1)
- Applications of Uv VisibleDocument3 pagesApplications of Uv VisibleLaiba ZulfiqarNo ratings yet
- UV-Vis InstrumentDocument7 pagesUV-Vis InstrumentNorizzatul Akmal100% (1)
- Analytical ChemistryDocument14 pagesAnalytical ChemistryHarrizul Rivai100% (1)
- Uv Visible Spectroscopy: by Nandesh V. PingaleDocument38 pagesUv Visible Spectroscopy: by Nandesh V. PingaleMohammed Adil ShareefNo ratings yet
- ElectrophoresisDocument7 pagesElectrophoresisnavedNo ratings yet
- Principles of IR SpectrosDocument6 pagesPrinciples of IR SpectrosSHEHA NUR AZZAHRA IBRAHIM -No ratings yet
- ICP-AES and ICP-MSDocument64 pagesICP-AES and ICP-MSSiska Winti Sone100% (1)
- Uv Visible SpectrosDocument50 pagesUv Visible SpectrosVacker Guzel50% (2)
- LC MsDocument24 pagesLC MsVshn VardhnNo ratings yet
- Uv Visible SpectrosDocument28 pagesUv Visible Spectrosjoshishravan3003No ratings yet
- NMRDocument142 pagesNMRMaulana Senjaya SusiloNo ratings yet
- Electrochemical Cells - New Advances in Fundamental Researches and ApplicatDocument250 pagesElectrochemical Cells - New Advances in Fundamental Researches and ApplicatRohit VermaNo ratings yet
- Analytical Chemistry Basic ConceptsDocument12 pagesAnalytical Chemistry Basic ConceptsNino Jay FabrosNo ratings yet
- Applied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistryDocument33 pagesApplied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistrySukh SindhiNo ratings yet
- Analytical Chemistry in ForensicsDocument7 pagesAnalytical Chemistry in ForensicsSergio Bugosen TannousNo ratings yet
- ChromatographyDocument28 pagesChromatographyyashaviNo ratings yet
- 02 KF TheoryDocument33 pages02 KF TheoryWina MarthaliaNo ratings yet
- Classification of Analytical MethodsDocument14 pagesClassification of Analytical MethodsNishchaya SinghNo ratings yet
- Role of Analytical Chemists in Industry - 2Document22 pagesRole of Analytical Chemists in Industry - 2hemakumarbNo ratings yet
- VoltammetryDocument38 pagesVoltammetryAndiswa pato100% (1)
- Analytical Chemistry UnfinishedDocument3 pagesAnalytical Chemistry UnfinishedJobeth Presto AlonzoNo ratings yet
- IR Spectra AnalysisDocument37 pagesIR Spectra AnalysisdevoydouglasNo ratings yet
- Basics of InstrumentationDocument35 pagesBasics of InstrumentationcoolhemakumarNo ratings yet
- Chromatography 2016Document42 pagesChromatography 2016IndraNo ratings yet
- Instrumental Analysis Lecture Notes IIDocument56 pagesInstrumental Analysis Lecture Notes IIcoolhemakumar100% (3)
- Electron Spin ResonanceDocument29 pagesElectron Spin Resonancecoolhemakumar100% (2)
- Basics of Dynamic ElectrochemistryDocument15 pagesBasics of Dynamic ElectrochemistryNaresh Chavan50% (2)
- Chapter 2: Chemicals, Apparatus, and Unit Operations of Analytical ChemistryDocument28 pagesChapter 2: Chemicals, Apparatus, and Unit Operations of Analytical Chemistryangela100% (2)
- Mass Spectrometry InstrumentationDocument3 pagesMass Spectrometry InstrumentationSangeetha priya SNo ratings yet
- Biosensor 7937718 PowerpointDocument39 pagesBiosensor 7937718 PowerpointHasanNo ratings yet
- LC MSDocument15 pagesLC MSSriNo ratings yet
- Analytical ChemistryDocument14 pagesAnalytical ChemistryDrMd Idris100% (2)
- Forensic ToxicologyDocument11 pagesForensic ToxicologySeptiandry Ade PutraNo ratings yet
- Intro To Electroanalytical ChemistryDocument38 pagesIntro To Electroanalytical Chemistryarun231187No ratings yet
- ColorimetryDocument21 pagesColorimetryM.PRASAD NAIDUNo ratings yet
- Electrophoresis and Capillary Electrophoresis PDFDocument21 pagesElectrophoresis and Capillary Electrophoresis PDFVinay kumarNo ratings yet
- An Introduction To Instrumental Methods PDFDocument4 pagesAn Introduction To Instrumental Methods PDFCharlot NavarroNo ratings yet
- NMR - A Non Destructive Food Evaluation Technique: Ramesh. VDocument44 pagesNMR - A Non Destructive Food Evaluation Technique: Ramesh. VAnkit GoyalNo ratings yet
- Mass SpectDocument28 pagesMass Spectapi-3723327100% (1)
- Mass SpectrosDocument111 pagesMass SpectrosMohammed75% (8)
- AAS AES Lambert Beer UCB008Document80 pagesAAS AES Lambert Beer UCB008Gagandeep ReehalNo ratings yet
- Ir SpectrometersDocument10 pagesIr SpectrometersVinícius FavrettoNo ratings yet
- Combustion MethodDocument13 pagesCombustion MethodAnaidEspinozaNo ratings yet
- 15 Spektro 01 Aas and AesDocument81 pages15 Spektro 01 Aas and AesmaudyNo ratings yet
- ASTDR - Toxicological Profil of CadmiumDocument487 pagesASTDR - Toxicological Profil of CadmiumpangkoklunNo ratings yet
- Industrial Chemistry - Syllabus Notes - Daniel WilsonDocument15 pagesIndustrial Chemistry - Syllabus Notes - Daniel WilsonPosclutoNo ratings yet
- NMRDocument35 pagesNMRnikhila11reddyNo ratings yet
- Basic OECD Princples of GLP by WHODocument30 pagesBasic OECD Princples of GLP by WHOastro79No ratings yet
- HPTLC-High Performance Thin Layer Chromatography: - Piyush L. Agrawal M.pharm - (Q.A.)Document39 pagesHPTLC-High Performance Thin Layer Chromatography: - Piyush L. Agrawal M.pharm - (Q.A.)Piyush Agrawal100% (2)
- An Introduction To Instrumental Methods PDFDocument4 pagesAn Introduction To Instrumental Methods PDFReagan Go100% (1)
- Flame PhotometerDocument18 pagesFlame PhotometerKuzhandai VeluNo ratings yet
- Atomic Emission SpectrosDocument5 pagesAtomic Emission SpectrosBea Uy0% (1)
- Fundamentals UV VISDocument53 pagesFundamentals UV VISRajesh Kumar Jha100% (1)
- IR SpectrosDocument60 pagesIR SpectrosdeepakNo ratings yet
- Essays on Analytical Chemistry: In Memory of Professor Anders RingbomFrom EverandEssays on Analytical Chemistry: In Memory of Professor Anders RingbomErkki WänninenNo ratings yet
- Oscillometry and Conductometry: International Series of Monographs on Analytical ChemistryFrom EverandOscillometry and Conductometry: International Series of Monographs on Analytical ChemistryNo ratings yet
- Colorimetry: Absorbance Is Directly Proportional To Concentration of FeDocument38 pagesColorimetry: Absorbance Is Directly Proportional To Concentration of Fedhekle_dNo ratings yet
- 2.visible and Ultraviolet (Uv) Light SpectrophotometerDocument65 pages2.visible and Ultraviolet (Uv) Light SpectrophotometerChabala Ng'anduNo ratings yet
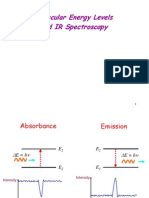
- Molecular Energy Levels and IR SpectrosDocument27 pagesMolecular Energy Levels and IR SpectroscoolhemakumarNo ratings yet
- Virtual Instrumentation ArchitectureDocument25 pagesVirtual Instrumentation Architecturecoolhemakumar100% (8)
- LabVIEW IntroductionDocument89 pagesLabVIEW Introductioncoolhemakumar100% (1)
- Digital and Pulse-Train Conditioning: Digital I/O InterfacingDocument6 pagesDigital and Pulse-Train Conditioning: Digital I/O InterfacingcoolhemakumarNo ratings yet
- Mass Spectrometry - Short NoteDocument2 pagesMass Spectrometry - Short NotecoolhemakumarNo ratings yet
- Displacement and PositionDocument6 pagesDisplacement and PositioncoolhemakumarNo ratings yet
- Daniel Gabriel Fahrenheit Thermometer Mercury Glass TemperatureDocument10 pagesDaniel Gabriel Fahrenheit Thermometer Mercury Glass TemperaturecoolhemakumarNo ratings yet
- Labview TutorialDocument609 pagesLabview Tutorialpeter3099No ratings yet
- CASE ToolsDocument5 pagesCASE ToolscoolhemakumarNo ratings yet
- Viscosity ConsistencyDocument8 pagesViscosity ConsistencycoolhemakumarNo ratings yet
- Industrial Instrumentation PDFDocument17 pagesIndustrial Instrumentation PDFSandhya NatarajanNo ratings yet
- VibrationDocument15 pagesVibrationYusman EkaNo ratings yet
- Thelma G. Manning and Zafar Iqbal - Polymeric Nitrogen Stabilized On Carbon Nanotubes: A Highly Energetic, Green ExplosiveDocument13 pagesThelma G. Manning and Zafar Iqbal - Polymeric Nitrogen Stabilized On Carbon Nanotubes: A Highly Energetic, Green ExplosiveYsam2No ratings yet
- C - Forano - Recent Trends in Electrochmical Detection of Phosphate in Actual Waters - 2018Document9 pagesC - Forano - Recent Trends in Electrochmical Detection of Phosphate in Actual Waters - 2018박지훈No ratings yet
- Platina ElektrolizaDocument27 pagesPlatina ElektrolizaGmmmm123No ratings yet
- Annurev Anchem 012809 102211Document25 pagesAnnurev Anchem 012809 102211Faran TAHIRNo ratings yet
- Sensors and Actuators B: Chemical: New Label Free CA125 Detection Based On Gold Nanostructured Screen-Printed ElectrodeDocument7 pagesSensors and Actuators B: Chemical: New Label Free CA125 Detection Based On Gold Nanostructured Screen-Printed ElectrodewardaninurindahNo ratings yet
- Preparation and Application of Platinum Composite Microelectrode (PCM) For The Routine Analysis of Acetaminophen in Pharmaceutical ProductsDocument7 pagesPreparation and Application of Platinum Composite Microelectrode (PCM) For The Routine Analysis of Acetaminophen in Pharmaceutical ProductsSafarudin AhmadNo ratings yet
- Electrochimica ActaDocument9 pagesElectrochimica ActaaaNo ratings yet
- Guide To Reliable PH MeasurementsDocument20 pagesGuide To Reliable PH MeasurementsYuri Jesus V.No ratings yet
- Jacob BondeDocument137 pagesJacob BondeĐặng Vũ Hoàng ĐứcNo ratings yet
- Amperometry: Working PrincipleDocument10 pagesAmperometry: Working PrincipleAbdulbar kelilNo ratings yet
- Anodic Stripping Voltammetry: BY: Chitrakshi GoelDocument32 pagesAnodic Stripping Voltammetry: BY: Chitrakshi GoelChitrakshi GoelNo ratings yet
- Addis Ababa University College of Health Sciences School of Pharmacy Department of Pharmaceutical Chemistry and PharmacognosyDocument35 pagesAddis Ababa University College of Health Sciences School of Pharmacy Department of Pharmaceutical Chemistry and PharmacognosyfahrillahnirmaNo ratings yet
- Double Potential-Step Chronoamperometry and Chronocoulometry at An Ultramicrodisk Electrode: Theory and ExperimentDocument8 pagesDouble Potential-Step Chronoamperometry and Chronocoulometry at An Ultramicrodisk Electrode: Theory and ExperimentMehanathan Maggie MikeyNo ratings yet
- Prism2009 Abstract BookDocument238 pagesPrism2009 Abstract Bookvdiazsuarez100% (1)
- J. J. Davis, K. S. Coleman Et Al.: 2003 Wiley-Vch Verlag GMBH & Co. Kgaa, Weinheim Chem. Eur. J. 2003, 9, 3732 3739Document8 pagesJ. J. Davis, K. S. Coleman Et Al.: 2003 Wiley-Vch Verlag GMBH & Co. Kgaa, Weinheim Chem. Eur. J. 2003, 9, 3732 3739Rana Sabouni TabariNo ratings yet
- Lecture 8 Electro-KineticsDocument63 pagesLecture 8 Electro-KineticsKhaled AbeedNo ratings yet
- Section I. Analytical Chemistry: Problem 1Document21 pagesSection I. Analytical Chemistry: Problem 1hakuna mata taNo ratings yet
- Cyclic Voltammetry Experiment: JamesDocument5 pagesCyclic Voltammetry Experiment: Jameshongluc1991100% (1)
- Free Download Laboratory Methods in Dynamic Electroanalysis M Teresa Fernandez Abedul Editor Full Chapter PDFDocument51 pagesFree Download Laboratory Methods in Dynamic Electroanalysis M Teresa Fernandez Abedul Editor Full Chapter PDFsean.crum166100% (11)
- Bismuto ExperienciaDocument4 pagesBismuto ExperienciaguiburNo ratings yet
- Copper - Anticancer - DPA Deriv - B - Art:10.1007/s11243-009-9200-5Document9 pagesCopper - Anticancer - DPA Deriv - B - Art:10.1007/s11243-009-9200-5skattejagNo ratings yet
- 2012 Dep of Conductivity On Charge Density and Echem Pot in Polymer SC W Ionic LiquidsDocument10 pages2012 Dep of Conductivity On Charge Density and Echem Pot in Polymer SC W Ionic LiquidsChris SmithNo ratings yet
- Electrodeposition of Conducting Transition Metal Oxide/Hydroxide Films From Aqueous SolutionDocument4 pagesElectrodeposition of Conducting Transition Metal Oxide/Hydroxide Films From Aqueous SolutionusercmdmcNo ratings yet
- 2021 - Abusaif - New Carbazole-Based Organic Dyes With Different Acceptors For Dye-SensitizedDocument11 pages2021 - Abusaif - New Carbazole-Based Organic Dyes With Different Acceptors For Dye-SensitizedTomas Delgado MontielNo ratings yet
- Characterizing Materials With Cyclic VoltammetryDocument9 pagesCharacterizing Materials With Cyclic VoltammetryanandramNo ratings yet
- Analytical ChemistryDocument6 pagesAnalytical ChemistryRodney Salazar0% (1)
- Core-Shell Prussian Blue Analogs With CompositionaDocument9 pagesCore-Shell Prussian Blue Analogs With CompositionaAkshay Rakesh Devikar AP19135030002No ratings yet