You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5814)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (844)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Disciplines and Ideas in Social SciencesDocument11 pagesDisciplines and Ideas in Social SciencesArjane Grace SullanoNo ratings yet
- Sop Iit KDocument2 pagesSop Iit KAbdul Gani0% (1)
- Daily Lesson Log School: Cadaloria High School Grade Level: 9 Teacher Teaching Dates Quarter Teaching Time SectionDocument3 pagesDaily Lesson Log School: Cadaloria High School Grade Level: 9 Teacher Teaching Dates Quarter Teaching Time SectionRichwellPanganibanSoliven100% (1)
- Reviewon Cervical CancerDocument7 pagesReviewon Cervical CancerAlmas TNo ratings yet
- Diabetes Mellitus: A Review On Pathophysiology, Current Status of Oral Medications and Future PerspectivesDocument22 pagesDiabetes Mellitus: A Review On Pathophysiology, Current Status of Oral Medications and Future PerspectivesAlmas TNo ratings yet
- Total Phenolic and Flavonoid Contents and Antioxidant Activities of Two Raphanus Sativus L. Cultivars (Cherry Belle and Valentine)Document6 pagesTotal Phenolic and Flavonoid Contents and Antioxidant Activities of Two Raphanus Sativus L. Cultivars (Cherry Belle and Valentine)Almas TNo ratings yet
- Raphanus Raphanistrum Subsp. Sativus: Scientifi C Name FamilyDocument41 pagesRaphanus Raphanistrum Subsp. Sativus: Scientifi C Name FamilyAlmas TNo ratings yet
- Art:10.1186/s13728 014 0020 7Document7 pagesArt:10.1186/s13728 014 0020 7Almas TNo ratings yet
- Rizki - Amalia - Faktor Faktor Perbesaran Prostat PDFDocument8 pagesRizki - Amalia - Faktor Faktor Perbesaran Prostat PDFAlmas TNo ratings yet
- Free Radical Biology & Medicine: Justin L. Rains, Sushil K. JainDocument9 pagesFree Radical Biology & Medicine: Justin L. Rains, Sushil K. JainAlmas TNo ratings yet
- Diabetes Insipidus HarrisonDocument18 pagesDiabetes Insipidus HarrisonAlmas TNo ratings yet
- FISH Book ReviewDocument25 pagesFISH Book Reviewshubs844926100% (3)
- Introduction To Society, Community and Education Dr. Mike Kelvin Nicole N. Buted at The End of This Module, Pre-Service Teachers Should Be Able ToDocument7 pagesIntroduction To Society, Community and Education Dr. Mike Kelvin Nicole N. Buted at The End of This Module, Pre-Service Teachers Should Be Able ToName S. CruzNo ratings yet
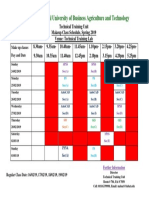
- Makeup Class Schedule - TTUDocument1 pageMakeup Class Schedule - TTUmehedeNo ratings yet
- P B N M: Media and Information LiteracyDocument11 pagesP B N M: Media and Information LiteracyJohn Stephen BagaygayNo ratings yet
- Midterm Exam UcspDocument3 pagesMidterm Exam UcspRicky Canico ArotNo ratings yet
- Analytical Study of Intrauterine Fetal Death Cases and Associated Maternal ConditionsDocument5 pagesAnalytical Study of Intrauterine Fetal Death Cases and Associated Maternal ConditionsNurvita WidyastutiNo ratings yet
- Valuation of AirThreadConnectionsDocument3 pagesValuation of AirThreadConnectionsmksscribd100% (1)
- Action Plan in MAPEH 2019 2020Document3 pagesAction Plan in MAPEH 2019 2020Maribel Dizon Mangio60% (5)
- Architecture in Shaping Child PsychologyDocument27 pagesArchitecture in Shaping Child Psychologychaturmukh97% (69)
- Aptis Listening Lv3 PT1 QBDocument2 pagesAptis Listening Lv3 PT1 QBNgọc Hân TrầnNo ratings yet
- Diaz - Meaning Beyond Words Analysis of Afrocuban Bata Drumming 2019Document180 pagesDiaz - Meaning Beyond Words Analysis of Afrocuban Bata Drumming 2019vxlaNo ratings yet
- Business FunctionDocument3 pagesBusiness Functionjithendar_thakurNo ratings yet
- Sampling & Sampling Distributions: Sanjay Rastogi, IIFT, New DelhiDocument16 pagesSampling & Sampling Distributions: Sanjay Rastogi, IIFT, New DelhiSanchi MongaNo ratings yet
- Sihem JebariDocument10 pagesSihem JebariSihem JebariNo ratings yet
- Aguado RondoDocument104 pagesAguado Rondoshanika123100% (1)
- SummaryDocument4 pagesSummaryMenora KazaryanNo ratings yet
- Slip Test - 1: Chapter - I: Nutritions: I. Answer The Following Question 1 4 4MDocument20 pagesSlip Test - 1: Chapter - I: Nutritions: I. Answer The Following Question 1 4 4MBaji Janjanam (CHANAKYA)No ratings yet
- Independent T Test LectureDocument27 pagesIndependent T Test LecturefatimahNo ratings yet
- Electronic Journal of Vedic StudiesDocument23 pagesElectronic Journal of Vedic StudiesJahangir Muhammad ArifNo ratings yet
- Excerpt From "The Loudest Voice in The Room: How The Brilliant, Bombastic Roger Ailes Built Fox News - and Divided A Country" by Gabriel Sherman.Document10 pagesExcerpt From "The Loudest Voice in The Room: How The Brilliant, Bombastic Roger Ailes Built Fox News - and Divided A Country" by Gabriel Sherman.OnPointRadioNo ratings yet
- The Last Lesson: By: Alphonse DaudetDocument10 pagesThe Last Lesson: By: Alphonse DaudetRohanNo ratings yet
- Dosti Foundation: Munir Ahmad, MD Chair Jessica Weinberg, JD, Publicity and Communications Coordinator 419-535-3214Document33 pagesDosti Foundation: Munir Ahmad, MD Chair Jessica Weinberg, JD, Publicity and Communications Coordinator 419-535-3214John KrochmalnyNo ratings yet
- DVS 2203 - 05 Testing of Welded Joints of Thermoplastics Plates and Tubes Technological Bend TestDocument3 pagesDVS 2203 - 05 Testing of Welded Joints of Thermoplastics Plates and Tubes Technological Bend TestAhmed GomaaNo ratings yet
- Dispute Settlement Machanism of WtoDocument17 pagesDispute Settlement Machanism of WtoShikha Gupta100% (1)
- Competence in Competency-Based Supervision PracticeDocument10 pagesCompetence in Competency-Based Supervision PracticeENSNo ratings yet
- GIS Data StructuresDocument48 pagesGIS Data Structureskrish_mplNo ratings yet
- People vs. Mariano - G.R. No. L-40527Document2 pagesPeople vs. Mariano - G.R. No. L-40527Karla ßallesteros100% (4)