You might also like
- Solution Reif Cap6 7Document13 pagesSolution Reif Cap6 7Marcio Particheli100% (1)
- Ai Ca3 NewDocument21 pagesAi Ca3 NewNavdeep SinghNo ratings yet
- Nonlinear Dynamics-And-Chaos-StrogatzDocument9 pagesNonlinear Dynamics-And-Chaos-Strogatzali.sahebiNo ratings yet
- Indistinguishable ParticlesDocument4 pagesIndistinguishable Particlesfwegfw3No ratings yet
- The Gibbs Statistical MechanicsDocument13 pagesThe Gibbs Statistical MechanicsIrvan PrakosoNo ratings yet
- Statistical Physics Problem Set SolutionsDocument12 pagesStatistical Physics Problem Set SolutionsJerry ScottNo ratings yet
- SLecture 7Document6 pagesSLecture 7Singh AnujNo ratings yet
- Chapter 3Document14 pagesChapter 3StefanPerendijaNo ratings yet
- hw3 2020 PDFDocument3 pageshw3 2020 PDFY LNo ratings yet
- CHEM 327 (Quantum Chemistry-I) Homework 2 (Due Date: 20/10/2023)Document2 pagesCHEM 327 (Quantum Chemistry-I) Homework 2 (Due Date: 20/10/2023)Eren KayaNo ratings yet
- Quatum Markov ChainDocument18 pagesQuatum Markov Chaink_zen865412No ratings yet
- Statistical Mechanics Chapter OverviewDocument15 pagesStatistical Mechanics Chapter OverviewSadhin SaleemNo ratings yet
- Stat Mech L1Document93 pagesStat Mech L1Lâm Văn Sa HuỳnhNo ratings yet
- Statistical Physics: Maxwell-Boltzmann DistributionDocument7 pagesStatistical Physics: Maxwell-Boltzmann Distributionst stNo ratings yet
- Monte Carlo Simulation of 2-D Ising Model Using Wang-Landau MethodDocument4 pagesMonte Carlo Simulation of 2-D Ising Model Using Wang-Landau Methodt_sairamNo ratings yet
- Stat Mech Overview Sci2Document5 pagesStat Mech Overview Sci2jdxdjdxdNo ratings yet
- Problem Set 1Document2 pagesProblem Set 1Uday RameshNo ratings yet
- Histogram Methods in Monte Carlo Simulations: BelliappaDocument61 pagesHistogram Methods in Monte Carlo Simulations: BelliappabelibeeNo ratings yet
- Ensemble TheoryDocument3 pagesEnsemble Theorymehdiramezani200No ratings yet
- Lecture 9Document7 pagesLecture 9Milind BhatiaNo ratings yet
- Barriers in The P-Spin Interacting Spin-Glass Model. The Dynamical ApproachDocument4 pagesBarriers in The P-Spin Interacting Spin-Glass Model. The Dynamical ApproachFahleza RejaNo ratings yet
- OU Open University SM358 2007 Exam SolutionsDocument23 pagesOU Open University SM358 2007 Exam Solutionssam smithNo ratings yet
- Elementos de Termodinamica EstadisticaDocument8 pagesElementos de Termodinamica EstadisticaCesar GalindoNo ratings yet
- Emtl Unit1 PDFDocument49 pagesEmtl Unit1 PDFnelapatikoteswarammaNo ratings yet
- This Homework Is Due On Wednesday, February 19, 2020, at 11:59PM. Self-Grades Are Due On Monday, February 24, 2019, at 11:59PM. 1 Complex NumbersDocument18 pagesThis Homework Is Due On Wednesday, February 19, 2020, at 11:59PM. Self-Grades Are Due On Monday, February 24, 2019, at 11:59PM. 1 Complex NumbersBella ZionNo ratings yet
- N - Body Problem: L1 A3-401 - Newton's Law of GravitationDocument22 pagesN - Body Problem: L1 A3-401 - Newton's Law of Gravitationudayang5330No ratings yet
- Boltzmann Entropy and The Microcanonical Ensemble: J. NaudtsDocument8 pagesBoltzmann Entropy and The Microcanonical Ensemble: J. Naudtsnguyen2ahoa3871No ratings yet
- Simulate The Electrostatic FieldDocument7 pagesSimulate The Electrostatic FieldxycNo ratings yet
- 2.1 The Canonical EnsembleDocument15 pages2.1 The Canonical EnsembleJesus MoralNo ratings yet
- Michelle Bodnar, Andrew Lohr May 5, 2017Document11 pagesMichelle Bodnar, Andrew Lohr May 5, 2017Mm AANo ratings yet
- Many Cheerful FactsDocument17 pagesMany Cheerful Factsbobmiller94No ratings yet
- Single Degree of Freedom System SDOF (Undamped)Document15 pagesSingle Degree of Freedom System SDOF (Undamped)Fahad ChaudharyNo ratings yet
- Fixed: E V N Morh orDocument9 pagesFixed: E V N Morh orbgiangre8372No ratings yet
- Single Degree of Freedom System (Undamped) : Energy MethodDocument15 pagesSingle Degree of Freedom System (Undamped) : Energy MethodVjh GNo ratings yet
- Statistical Physics (MSC Phy) 4Document7 pagesStatistical Physics (MSC Phy) 4SACHIN VISHWAKARMANo ratings yet
- Generalized Riccati Equation and Spectral Factorization For Discrete-Time Descriptor SystemDocument4 pagesGeneralized Riccati Equation and Spectral Factorization For Discrete-Time Descriptor SystemsumathyNo ratings yet
- SHM Review - SHM Using Differential Equations - Critical DampingDocument18 pagesSHM Review - SHM Using Differential Equations - Critical Dampingaaljuhani123No ratings yet
- Fundamental Theorem of Curves and Non-Unit Speed CurvesDocument7 pagesFundamental Theorem of Curves and Non-Unit Speed CurvesJean Pierre RukundoNo ratings yet
- 1 s2 2.0 S0362546X1100633X MainDocument9 pages1 s2 2.0 S0362546X1100633X Mainsatitz chongNo ratings yet
- Statistical Thermodynamics Ensemble DistributionsDocument3 pagesStatistical Thermodynamics Ensemble DistributionsEmmanuel JoyNo ratings yet
- The Second Law of Thermodynamics Energy ExchangeDocument30 pagesThe Second Law of Thermodynamics Energy Exchangekarimi-15No ratings yet
- Coordinate Systems: 1 System DefinitionsDocument3 pagesCoordinate Systems: 1 System DefinitionsRizky FitriansyahNo ratings yet
- Physics 101B. Modern Physics. Professor Dine: 1 Stirling's FormulaDocument2 pagesPhysics 101B. Modern Physics. Professor Dine: 1 Stirling's Formulajijay110694No ratings yet
- Mathematical Association of AmericaDocument4 pagesMathematical Association of America23SquareNo ratings yet
- Small Excitonic Complexes in A Disk-Shaped Quantum Dot: Ricardo Perez, Augusto GonzalezDocument8 pagesSmall Excitonic Complexes in A Disk-Shaped Quantum Dot: Ricardo Perez, Augusto GonzalezJairofisico JaramilloNo ratings yet
- Booklet 2012Document69 pagesBooklet 2012Esteban RodríguezNo ratings yet
- Fortescue1918 1 50Document50 pagesFortescue1918 1 50CARLOS ALBERTO GOMEZ REYESNo ratings yet
- Possible Questions Exam Non-Equilibrium Statistical MechanicsDocument4 pagesPossible Questions Exam Non-Equilibrium Statistical MechanicsEgop3105No ratings yet
- 7-Ensembles 0Document20 pages7-Ensembles 0loop arrayNo ratings yet
- Strong Convergence Theorems For Strict Pseudocontractions in Uniformly Convex Banach SpacesDocument9 pagesStrong Convergence Theorems For Strict Pseudocontractions in Uniformly Convex Banach SpacesnesmaeilnNo ratings yet
- Lab 3Document20 pagesLab 3alif fudenNo ratings yet
- Solutions To Problem Set 2: P (E) + DPDocument12 pagesSolutions To Problem Set 2: P (E) + DPlantea1No ratings yet
- ME421-Single Degree of Freedom System (Undamped) - v2 PDFDocument13 pagesME421-Single Degree of Freedom System (Undamped) - v2 PDFAnonymous eeMcgmX3WNo ratings yet
- Canonical Partition Function DerivationDocument9 pagesCanonical Partition Function DerivationJay SteeleNo ratings yet
- Modeling Transfer Functions in ScilabDocument17 pagesModeling Transfer Functions in ScilabClinth JhonNo ratings yet
- Flash Calc 3Document16 pagesFlash Calc 3Mohamed MamdouhNo ratings yet
- Reaction Rate Theories and CatalysisDocument7 pagesReaction Rate Theories and CatalysisJosephine ChenNo ratings yet
- Booklet PDFDocument73 pagesBooklet PDFESHETUNo ratings yet
- Real Variables with Basic Metric Space TopologyFrom EverandReal Variables with Basic Metric Space TopologyRating: 5 out of 5 stars5/5 (1)
- Phys 221 ExercisesDocument4 pagesPhys 221 ExercisesKristine Rodriguez-CarnicerNo ratings yet
- Derive The The Generalized Partition Function For Fermions (Fermi-Dirac Particles) and Bosons (Bose-Einstein Particles)Document5 pagesDerive The The Generalized Partition Function For Fermions (Fermi-Dirac Particles) and Bosons (Bose-Einstein Particles)Kristine Rodriguez-CarnicerNo ratings yet
- Phys - 221 - PROBLEM SET #1Document4 pagesPhys - 221 - PROBLEM SET #1Kristine Rodriguez-CarnicerNo ratings yet
- Two-particle system heat capacityDocument2 pagesTwo-particle system heat capacityKristine Rodriguez-CarnicerNo ratings yet
- Derive The The Generalized Partition Function For Fermions (Fermi-Dirac Particles) and Bosons (Bose-Einstein Particles)Document5 pagesDerive The The Generalized Partition Function For Fermions (Fermi-Dirac Particles) and Bosons (Bose-Einstein Particles)Kristine Rodriguez-CarnicerNo ratings yet
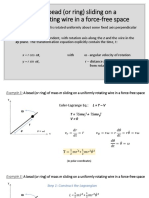
- Example 3: A Bead (Or Ring) Sliding On A: Uniformly Rotating Wire in A Force-Free SpaceDocument8 pagesExample 3: A Bead (Or Ring) Sliding On A: Uniformly Rotating Wire in A Force-Free SpaceKristine Rodriguez-Carnicer100% (1)
- Light Interference From A Soap Film: A Revisited Quasi-Monochromatic ExperimentDocument20 pagesLight Interference From A Soap Film: A Revisited Quasi-Monochromatic ExperimentKristine Rodriguez-CarnicerNo ratings yet
- Light Interference From A Soap Film: A Revisited Quasi-Monochromatic ExperimentDocument20 pagesLight Interference From A Soap Film: A Revisited Quasi-Monochromatic ExperimentKristine Rodriguez-CarnicerNo ratings yet
- Floyd - Vectors NotesDocument41 pagesFloyd - Vectors NotesKristine Rodriguez-CarnicerNo ratings yet
- Asian Monsoon System RRL TableDocument5 pagesAsian Monsoon System RRL TableKristine Rodriguez-CarnicerNo ratings yet
- Debris Flow SimulationDocument42 pagesDebris Flow SimulationKristine Rodriguez-CarnicerNo ratings yet
- Problem #2.1 Brachistochrone With VoDocument2 pagesProblem #2.1 Brachistochrone With VoKristine Rodriguez-CarnicerNo ratings yet
- Energy Function: Gabriela Gonz Alez September 9, 2006Document2 pagesEnergy Function: Gabriela Gonz Alez September 9, 2006Kristine Rodriguez-CarnicerNo ratings yet
- D'Alembert's Principle and Lagrange's EquationDocument10 pagesD'Alembert's Principle and Lagrange's EquationKristine Rodriguez-CarnicerNo ratings yet
- TC Rainfall Contribution to Philippines RainfallDocument17 pagesTC Rainfall Contribution to Philippines RainfallKristine Rodriguez-CarnicerNo ratings yet
- Chap 3Document17 pagesChap 3Yessenia Madrid100% (1)
- Monsoon RRLDocument11 pagesMonsoon RRLKristine Rodriguez-CarnicerNo ratings yet
- The Second Law of Thermodynamics: Refrigerators EntropyDocument33 pagesThe Second Law of Thermodynamics: Refrigerators EntropyKristine Rodriguez-CarnicerNo ratings yet
- Sturm Liouville Euler LagrangeDocument22 pagesSturm Liouville Euler LagrangejojolilloNo ratings yet
- Os Manual (Arranged)Document78 pagesOs Manual (Arranged)P.DhivyaNo ratings yet
- MATHDocument10 pagesMATHSherylyn BanaciaNo ratings yet
- Torque Equation of Three Phase Induction Motor - Electrical4UDocument14 pagesTorque Equation of Three Phase Induction Motor - Electrical4Ukaustav choudhuryNo ratings yet
- Amc 8 2019Document10 pagesAmc 8 2019Madhu Mohan ChandranNo ratings yet
- Lte-Rf-Planning-Optimization Drive Testing Et AlDocument60 pagesLte-Rf-Planning-Optimization Drive Testing Et AlEfosa AigbeNo ratings yet
- Super Cheatsheet Deep LearningDocument13 pagesSuper Cheatsheet Deep LearningcidsantNo ratings yet
- Noc20-Cs28 Week 07 Assignment 02Document6 pagesNoc20-Cs28 Week 07 Assignment 02Praveen Urs RNo ratings yet
- Quezon Naguilian, IsabelaDocument2 pagesQuezon Naguilian, IsabelaYvette Marie Yaneza NicolasNo ratings yet
- Circular Motion Lab For Physics 2016 SpringDocument1 pageCircular Motion Lab For Physics 2016 SpringIrwansyah Ramadhani100% (1)
- ADS 1st Semester Course OutlinesDocument6 pagesADS 1st Semester Course OutlinesClaire MarshallNo ratings yet
- STATEMENT ASE(3)Document2 pagesSTATEMENT ASE(3)dacianzNo ratings yet
- Research - Grade 12Document4 pagesResearch - Grade 12JJ Angelo ChongNo ratings yet
- Expressing Large Numbers in Different FormsDocument63 pagesExpressing Large Numbers in Different FormsJoel ValdezNo ratings yet
- The Design of Vibro Replacement (Priebe Teory)Document17 pagesThe Design of Vibro Replacement (Priebe Teory)paduco100% (3)
- BT07 BMT17 Seismic Slope Displacement v5Document16 pagesBT07 BMT17 Seismic Slope Displacement v5Ravi SalimathNo ratings yet
- Smoothing Frequency Domain FiltersDocument22 pagesSmoothing Frequency Domain FiltersRaymond PraveenNo ratings yet
- The Science of Tone-Color: Exploring the Relationship Between Sound and LightDocument105 pagesThe Science of Tone-Color: Exploring the Relationship Between Sound and LightXeropointNo ratings yet
- I-Day 35Document3 pagesI-Day 35Rainman InsanityNo ratings yet
- Normal Distribution1Document24 pagesNormal Distribution1Arun RaiNo ratings yet
- Novel Fiber Optic Biosensors Based On Nanoplasmonic and Interferometric ModalitiesDocument152 pagesNovel Fiber Optic Biosensors Based On Nanoplasmonic and Interferometric ModalitiesIan MuriNo ratings yet
- BIP39 Word & Numbers ListDocument7 pagesBIP39 Word & Numbers Listcontadetestes321No ratings yet
- Chapter 5 Static Timing AnalysisDocument126 pagesChapter 5 Static Timing Analysisjeevan_rao100% (4)
- 10 Robotic Filament WindingDocument9 pages10 Robotic Filament WindingYohannes RegassaNo ratings yet
- Semester I Semester - Iv Semester - Vii: Total 18 4 8 Total 24 28 30Document119 pagesSemester I Semester - Iv Semester - Vii: Total 18 4 8 Total 24 28 30John ShugerNo ratings yet
- Grade 11 ABM and STEM ExamDocument9 pagesGrade 11 ABM and STEM ExamkeyedNo ratings yet
- Ye Swan Htet - Maths - Stage - 7 - 01Document16 pagesYe Swan Htet - Maths - Stage - 7 - 01Ye Swan HtetNo ratings yet
- MATH20602 - 2014 exam questionsDocument15 pagesMATH20602 - 2014 exam questionsMuhammad KamranNo ratings yet
- Diff Shortening of Tall Steel Building Columns TaranathDocument8 pagesDiff Shortening of Tall Steel Building Columns TaranathgoggingsNo ratings yet
- Hedge Fund Risk Appetite:: A Prospect Theory PerspectiveDocument29 pagesHedge Fund Risk Appetite:: A Prospect Theory Perspectivephuongapo9xNo ratings yet
- The A-Mazing RaceDocument16 pagesThe A-Mazing Raceapi-489665585No ratings yet