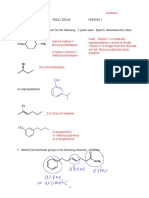
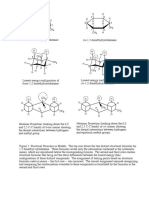
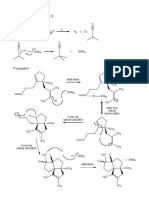
You might also like
- 4 Supplementary Exercise Acids and BasesDocument113 pages4 Supplementary Exercise Acids and Bases云吸仓鼠吉尼斯保持者No ratings yet
- Alkenes Reactions NotesDocument14 pagesAlkenes Reactions NotesMartin AlvinNo ratings yet
- MSDS MerkuriDocument6 pagesMSDS MerkuriRizkaAuliandini100% (1)
- Conformations of Organic MoleculesDocument21 pagesConformations of Organic MoleculesAkash KolliNo ratings yet
- Hydrogen and Its Compounds: ST THDocument8 pagesHydrogen and Its Compounds: ST THAravind NarasimhaluNo ratings yet
- TestFinal 350 v1 AnswersDocument8 pagesTestFinal 350 v1 AnswersJabe KoyNo ratings yet
- 1,3 CycloDocument1 page1,3 CycloIsmail ZitouniNo ratings yet
- R Vs S AnsDocument2 pagesR Vs S AnsAtul SinghNo ratings yet
- 7.7: Fischer Projections - Representation of A Three-DimensionalDocument3 pages7.7: Fischer Projections - Representation of A Three-DimensionalFakhri ElabbarNo ratings yet
- Krisna Dewi 1913031004Document33 pagesKrisna Dewi 1913031004NOVAL TAUHIDNo ratings yet
- Homework Problems: Structure, Bonding & Hybridization 1. The Molecule Shown Below Is Griseofulvin, An Antifungal CompoundDocument8 pagesHomework Problems: Structure, Bonding & Hybridization 1. The Molecule Shown Below Is Griseofulvin, An Antifungal CompoundPrachi KaushikNo ratings yet
- OrganicChemistryChapter7 PDFDocument30 pagesOrganicChemistryChapter7 PDFSeanne CruzNo ratings yet
- Ilovepdf MergedDocument50 pagesIlovepdf MergedAMARTYA 4863No ratings yet
- Alkynes and RadicalsDocument7 pagesAlkynes and RadicalsArmando Shehi SayhellotogoodbyeNo ratings yet
- Elimination Reactions: E1, E2Document11 pagesElimination Reactions: E1, E2Susobhan ghoshNo ratings yet
- Assignment 1Document12 pagesAssignment 1Tshiamo MotaungNo ratings yet
- ConformationDocument30 pagesConformationRithvik ShastryNo ratings yet
- Chapter 8-Reactions of AlkenesDocument35 pagesChapter 8-Reactions of Alkenes張湧浩No ratings yet
- CY2102Document2 pagesCY2102Prarabdha SharmaNo ratings yet
- Jee Main Full Syllabus Test-7-Solutions-1Document11 pagesJee Main Full Syllabus Test-7-Solutions-1ishaanpathak6No ratings yet
- Chapter 4Document20 pagesChapter 4Simran Saiinii0% (1)
- DM pp21-40Document20 pagesDM pp21-40MLUNGISI MkhwanaziNo ratings yet
- 3 15 Revision Guide NMRDocument5 pages3 15 Revision Guide NMRPraveenaNo ratings yet
- CHEM3411 Lab DehydrationDocument5 pagesCHEM3411 Lab DehydrationBianca AlexisNo ratings yet
- Madeleine Ceri - Final Exam CHE-A-02Document6 pagesMadeleine Ceri - Final Exam CHE-A-02Madeleine CeriNo ratings yet
- 3.15 Revision Guide NMRDocument5 pages3.15 Revision Guide NMRyimiyeh441No ratings yet
- Important Concepts: Conformational Analysis of AlkanesDocument4 pagesImportant Concepts: Conformational Analysis of AlkanesKayla JoezetteNo ratings yet
- Elimination Reactions: Elimination Reaction: A Reaction in Which A Molecule Loses Atoms or Groups of AtomsDocument8 pagesElimination Reactions: Elimination Reaction: A Reaction in Which A Molecule Loses Atoms or Groups of AtomsMohammed Adil ShareefNo ratings yet
- Addition ReactionsDocument54 pagesAddition ReactionsRithvik ShastryNo ratings yet
- Ejercicios Axial EcuatorialDocument1 pageEjercicios Axial EcuatorialJulio Cesar Boada MartinezNo ratings yet
- CH CH CH CH I: BRCH CH CH CCH BR CH CHDocument24 pagesCH CH CH CH I: BRCH CH CH CCH BR CH CHSam TabujaraNo ratings yet
- TABORADA - Act8 (Ketones and Aldehydes)Document4 pagesTABORADA - Act8 (Ketones and Aldehydes)Justin Habaña TaboradaNo ratings yet
- Answer Scheme ORGANIC For Set 2Document2 pagesAnswer Scheme ORGANIC For Set 2Hafizah HalimNo ratings yet
- Problem Set #6 AnswersDocument3 pagesProblem Set #6 AnswersStefan BamNo ratings yet
- Chapter 7 HaloalkanesDocument11 pagesChapter 7 HaloalkanesSandra JohnNo ratings yet
- 맥머리의 유기화학 9판 (답)Document29 pages맥머리의 유기화학 9판 (답)빅옹일No ratings yet
- H BR H H 1 2 1 2 (A, E) Cis-1,2-Dibromocyclohexane BR H H BR H BR 1 2 1 2 (A, A) Trans-1,2-Dibromocyclohexane BR BR BR HDocument19 pagesH BR H H 1 2 1 2 (A, E) Cis-1,2-Dibromocyclohexane BR H H BR H BR 1 2 1 2 (A, A) Trans-1,2-Dibromocyclohexane BR BR BR HVIGHNESH BALKRISHNA LOKARENo ratings yet
- 2906 Chemistry Paper With Solution EveningDocument7 pages2906 Chemistry Paper With Solution EveningPoojan ShahNo ratings yet
- RS Conformational AnalysisL4-L5.Ppt 1Document40 pagesRS Conformational AnalysisL4-L5.Ppt 1Vishal GarimellaNo ratings yet
- Reductions PPT 29-08-2020Document12 pagesReductions PPT 29-08-2020jkc collegeNo ratings yet
- Coursebook Answers Chapter 18 Asal ChemistryDocument4 pagesCoursebook Answers Chapter 18 Asal ChemistryMarin PesicNo ratings yet
- Alkene Reaction ConnectionsDocument1 pageAlkene Reaction ConnectionsMackay SteffensenNo ratings yet
- New CHY3201 Chapter 9 Addition ReactionDocument31 pagesNew CHY3201 Chapter 9 Addition Reaction222418No ratings yet
- Organic Reactions Summary Alkenes, Alkynes and Variations For Use As A Study Guide BeauchampDocument45 pagesOrganic Reactions Summary Alkenes, Alkynes and Variations For Use As A Study Guide BeauchampShaker MahmoodNo ratings yet
- Stereochemistry of OrganicDocument35 pagesStereochemistry of OrganicRams ChanderNo ratings yet
- 314 Stereochem ProbsDocument14 pages314 Stereochem ProbsAtul SinghNo ratings yet
- Acid - Base Equilibria: Prof. Dr. Hassan Sedeira Prof. Dr. Elham Y. HashemDocument13 pagesAcid - Base Equilibria: Prof. Dr. Hassan Sedeira Prof. Dr. Elham Y. HashemMoamen MohamedNo ratings yet
- Problem Set 3, Question 2 InitiationDocument1 pageProblem Set 3, Question 2 InitiationRudra Shankha NandyNo ratings yet
- Naming Alcohols PDFDocument1 pageNaming Alcohols PDFumairgul841No ratings yet
- Problem Set 9 KEYDocument3 pagesProblem Set 9 KEYCARLOS ALBERTO OSORIO MARTINEZNo ratings yet
- Nomenclature of Organic CompoundsDocument80 pagesNomenclature of Organic CompoundsSajjad MiraniNo ratings yet
- Problem Set 7 AnswersDocument6 pagesProblem Set 7 AnswersVasanthakumar.vNo ratings yet
- Organic Chemistry Chapter 14Document27 pagesOrganic Chemistry Chapter 14Đỗ Minh HuânNo ratings yet
- C Sol Ch-21 HydrocarbonsDocument8 pagesC Sol Ch-21 Hydrocarbonsmysoftinfo.incNo ratings yet
- Vide Notes and Remarks: Strictly Ima Mmi'sDocument6 pagesVide Notes and Remarks: Strictly Ima Mmi'sgodwin solomonNo ratings yet
- Exam1 S06 Answers PDFDocument6 pagesExam1 S06 Answers PDFIdrees Ah MalikNo ratings yet
- OrganicChemistryChapter5 PDFDocument19 pagesOrganicChemistryChapter5 PDFJuliet Tatiana CumbeNo ratings yet
- Chemistry I (Organic) : Stereochemistry: Diastereoisomers DR Alan Spivey Office: 834 C1 E-Mail: Tel.: 45841Document2 pagesChemistry I (Organic) : Stereochemistry: Diastereoisomers DR Alan Spivey Office: 834 C1 E-Mail: Tel.: 45841Francisco RomeroNo ratings yet
- Lecture 16 (AK 1)Document44 pagesLecture 16 (AK 1)vipulugaleNo ratings yet
- Chemistry (Full Test) - Paper 1 - SolutionsDocument6 pagesChemistry (Full Test) - Paper 1 - SolutionsRavi Kiran KoduriNo ratings yet
- alfa-3-N-acetil-D-alopiranosil - (1 3) - Beta-6-Desoxi-L-Psicofuranosil - (2 5) - D-LixofuranosaDocument1 pagealfa-3-N-acetil-D-alopiranosil - (1 3) - Beta-6-Desoxi-L-Psicofuranosil - (2 5) - D-LixofuranosaShaira Benavides0% (1)
- XXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973From EverandXXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973No ratings yet
- Characterization of Chitin, Chitosan by DSC PDFDocument9 pagesCharacterization of Chitin, Chitosan by DSC PDFtaufik ismullahNo ratings yet
- Haloalkanes and HaloarenesDocument14 pagesHaloalkanes and HaloarenesKalpesh BishnoiNo ratings yet
- The Management of Heat Stress Through NutritionDocument45 pagesThe Management of Heat Stress Through NutritionaaronjulesNo ratings yet
- Zmoq : Narjmwu H$Mos H$Mo Cîma NWPÑVH$M Ho$ - Wi N Ð Na Adí' (Bio & Amob ZDocument19 pagesZmoq : Narjmwu H$Mos H$Mo Cîma NWPÑVH$M Ho$ - Wi N Ð Na Adí' (Bio & Amob ZRavneet KaurNo ratings yet
- WEEK 1, Grade 10Document2 pagesWEEK 1, Grade 10Sheela BatterywalaNo ratings yet
- F F O P L: Insufine - VI 610Document2 pagesF F O P L: Insufine - VI 610Karishma PrabhuNo ratings yet
- FOODLAB Line Configurations PDFDocument9 pagesFOODLAB Line Configurations PDFMr. MeNo ratings yet
- Ideal Solns, Colig PropsDocument19 pagesIdeal Solns, Colig PropsBeverly BalunonNo ratings yet
- Infion 10 Juni 2020Document2 pagesInfion 10 Juni 2020Shane RNo ratings yet
- Levoshin 100 MLDocument1 pageLevoshin 100 MLSharfina Akter BithiNo ratings yet
- Surat Bukti Barang KeluarDocument3 pagesSurat Bukti Barang KeluarPuskesmas Dabun GelangNo ratings yet
- Formulation and Evaluation of Poly Herbal Body ScrubDocument6 pagesFormulation and Evaluation of Poly Herbal Body ScrubInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- MPN Untuk AlgaDocument8 pagesMPN Untuk AlgaIyak LectureNo ratings yet
- Sistemas Jiffy DmeDocument31 pagesSistemas Jiffy DmeGerman LagNo ratings yet
- Pyrogen Test and Its Determination Using Rabbits - Pharmaceutical GuidelinesDocument1 pagePyrogen Test and Its Determination Using Rabbits - Pharmaceutical Guidelinesstalker akoNo ratings yet
- DGU-403 DGU-405: Instruction ManualDocument30 pagesDGU-403 DGU-405: Instruction ManualCrystal LinNo ratings yet
- Compositional Changes of Crude Oil SARA Fractions Due To Biodegradation and Adsorption Supported On Colloidal Support Such As Clay Susing IatroscanDocument13 pagesCompositional Changes of Crude Oil SARA Fractions Due To Biodegradation and Adsorption Supported On Colloidal Support Such As Clay Susing IatroscanNatalia KovalovaNo ratings yet
- Catalogo Natureza 2020 EnglishDocument10 pagesCatalogo Natureza 2020 EnglishJaciara RibeiroNo ratings yet
- Product Identification Osbond 299Document2 pagesProduct Identification Osbond 299Sonia Rahima PutriNo ratings yet
- Methods For Measuring Ozone Concentration in Ozone-Treated WaterDocument3 pagesMethods For Measuring Ozone Concentration in Ozone-Treated WaterChanthol RibeiroNo ratings yet
- 56 1 2 ChemistryDocument19 pages56 1 2 ChemistryParth SaxenaNo ratings yet
- Gram StainingDocument5 pagesGram StainingDavid WolfyNo ratings yet
- Chloe Brylle S. RoldanDocument6 pagesChloe Brylle S. RoldanChloe RoldanNo ratings yet
- 2 Hydroxybenzoic Acid Colorimetry Assay Student PDFDocument2 pages2 Hydroxybenzoic Acid Colorimetry Assay Student PDFMj BalNo ratings yet
- The Effects of Nitrogen Fertilizer On Plant GrowthDocument7 pagesThe Effects of Nitrogen Fertilizer On Plant GrowthEditor IJTSRDNo ratings yet
- Alfa Laval Heating and Cooling Hub The Theory Behind Heat TransferDocument12 pagesAlfa Laval Heating and Cooling Hub The Theory Behind Heat TransferLuiz guilherme OliveiraNo ratings yet
- Rheological Properties of Some Thermotropic Liquid Crystalline PolymersDocument7 pagesRheological Properties of Some Thermotropic Liquid Crystalline PolymersAdityaNo ratings yet