You might also like
- PCR Lab ProtocolDocument5 pagesPCR Lab Protocolhk8atema1lNo ratings yet
- Advanced Pharmaceutical analysisFrom EverandAdvanced Pharmaceutical analysisRating: 4.5 out of 5 stars4.5/5 (2)
- NBME Step 2 Form 3Document45 pagesNBME Step 2 Form 3bostickdrew16100% (1)
- 3bsmt1 Bobier, Ashwelldonne Molbio PCRDocument9 pages3bsmt1 Bobier, Ashwelldonne Molbio PCRAshwell Donne BobierNo ratings yet
- 1 L Mcdonald More Than Just Little Men Women and Fat Loss 2698947813Document25 pages1 L Mcdonald More Than Just Little Men Women and Fat Loss 2698947813Carlos0% (1)
- Knowledge Is PowerDocument17 pagesKnowledge Is PowerRohitKumarSahu100% (1)
- Dna Purification and Extraction Practical ReportDocument8 pagesDna Purification and Extraction Practical ReportAnselmo ManishaNo ratings yet
- Hongkong v. Olalia, Jr. (CASE DIGEST)Document3 pagesHongkong v. Olalia, Jr. (CASE DIGEST)Samantha NicoleNo ratings yet
- Autodesk - 300 Records Profiling - 2nd Lot For ValidationDocument21 pagesAutodesk - 300 Records Profiling - 2nd Lot For ValidationVainiNo ratings yet
- Redox Indicators. Characteristics and ApplicationsFrom EverandRedox Indicators. Characteristics and ApplicationsRating: 5 out of 5 stars5/5 (1)
- 015 PCR Protocols 1st EditionDocument386 pages015 PCR Protocols 1st EditionmuyeedahmedNo ratings yet
- 08 - Chapter 2Document14 pages08 - Chapter 2The FrequencyNo ratings yet
- LSM1102 - Alkaline Lysis Original ArticleDocument11 pagesLSM1102 - Alkaline Lysis Original Articlegivena2ndchanceNo ratings yet
- Lab Report 1Document3 pagesLab Report 1Nurul NadiaNo ratings yet
- DNA Lab 1Document4 pagesDNA Lab 1Abdul Mueez LoneNo ratings yet
- 10 Enzymes For Modifying and Labeling DNA and RN - 1987 - Methods in EnzymologDocument17 pages10 Enzymes For Modifying and Labeling DNA and RN - 1987 - Methods in EnzymologMontsZs G-oNo ratings yet
- Laboratory Manual BTY312 Genetic EngineeringDocument11 pagesLaboratory Manual BTY312 Genetic EngineeringGeetanjali GorainNo ratings yet
- RNA - TRIZOL Extraction Lab ProtocolDocument6 pagesRNA - TRIZOL Extraction Lab ProtocolshubhambhauNo ratings yet
- Bimbo Im 1979Document12 pagesBimbo Im 1979Kasthuri Thirupathi100% (1)
- INTRODUCTION Isolation of Plant DNADocument23 pagesINTRODUCTION Isolation of Plant DNAkhushbujain7992No ratings yet
- Transformation ConfirmationDocument5 pagesTransformation ConfirmationMuhammad MoeezNo ratings yet
- Dna Isolation For Red AlgaeDocument7 pagesDna Isolation For Red AlgaeBalasankar ThangaswamyNo ratings yet
- DNA Extraction MethodsDocument6 pagesDNA Extraction MethodsTeflon SlimNo ratings yet
- Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform ExtractionDocument4 pagesSingle-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform ExtractionAnggraeni Arum SNo ratings yet
- Primer Extension, and Nuclease MappingDocument35 pagesPrimer Extension, and Nuclease Mappingamar7chauhanNo ratings yet
- CHEM4242-DNA RevisedDocument6 pagesCHEM4242-DNA Revisedjhui3894No ratings yet
- Rapid Non-Enzymatic Preparation HMW From: A DNA Blood For RFLP StudiesDocument1 pageRapid Non-Enzymatic Preparation HMW From: A DNA Blood For RFLP StudiesRaj KumarNo ratings yet
- 1991-One Step 'Miniprep' Method For The Isolation of PlasmidDocument1 page1991-One Step 'Miniprep' Method For The Isolation of Plasmidrasime.demirelNo ratings yet
- DBT-HRD Training ManualDocument87 pagesDBT-HRD Training ManualPiyush Ranjan BeheraNo ratings yet
- DNA Isolation, Restriction Digest, and ElectrophoresisDocument11 pagesDNA Isolation, Restriction Digest, and ElectrophoresisRosemarie Dawn Tagare100% (2)
- Functional GenomicsDocument7 pagesFunctional GenomicsHenrique MoraisNo ratings yet
- Waynehughes 1988Document5 pagesWaynehughes 1988AndreaMolinaDuránNo ratings yet
- 4) DNA ExtractionDocument11 pages4) DNA ExtractionajiesyahbarieNo ratings yet
- Laboratory Protocols: Plasmid DNA IsolationDocument7 pagesLaboratory Protocols: Plasmid DNA Isolationone kilometerNo ratings yet
- Handout For Workshop (DAY 01) (KSBT)Document7 pagesHandout For Workshop (DAY 01) (KSBT)Michael KahnwaldNo ratings yet
- The Isolation of High Molecular Weight Eukaryotic DNA: C. G. P. MathewDocument4 pagesThe Isolation of High Molecular Weight Eukaryotic DNA: C. G. P. MathewSujoy DebNo ratings yet
- Extraction of DNA From Whole BloodDocument5 pagesExtraction of DNA From Whole BloodvishankguptaNo ratings yet
- Extraccion de ADN HongosDocument4 pagesExtraccion de ADN HongosNelver MorenoNo ratings yet
- Inverse PCRDocument6 pagesInverse PCRHiromi UchimaNo ratings yet
- DNA IsolationDocument10 pagesDNA Isolationromaehab201912No ratings yet
- Southern Blot Lab ReportDocument10 pagesSouthern Blot Lab ReportPeter Ickes0% (1)
- 18BTC108J - Plant Biotech Lab - Exp. No. 3 and 4Document10 pages18BTC108J - Plant Biotech Lab - Exp. No. 3 and 4Chris PenielNo ratings yet
- Extraction of High Molecular Weight DNA From Eukaryotic Cells Molecular Biology Lab #7Document4 pagesExtraction of High Molecular Weight DNA From Eukaryotic Cells Molecular Biology Lab #7Mahnoor ArshadNo ratings yet
- Mini PrepDocument6 pagesMini PrepWilson GomargaNo ratings yet
- 08091003RNA ExtractionDocument3 pages08091003RNA ExtractionAnonymous WVPfUWwi3No ratings yet
- Bacterial DNA IsolationDocument6 pagesBacterial DNA IsolationVikram SinghNo ratings yet
- Comprobación de Extracción de Rna, Dna Cromosómico, Dna Plasmídico Por Espectrofotometria Y ElectroforesisDocument8 pagesComprobación de Extracción de Rna, Dna Cromosómico, Dna Plasmídico Por Espectrofotometria Y ElectroforesisJavier RodriguezNo ratings yet
- Lab 1Document8 pagesLab 1Gana KhaledNo ratings yet
- BSC Micro IV Sem PracticalsDocument11 pagesBSC Micro IV Sem PracticalsPrabhu SaxenaNo ratings yet
- RNA Extraction: OverviewDocument3 pagesRNA Extraction: OverviewArturo BasantezNo ratings yet
- Dna Extraction공부Document5 pagesDna Extraction공부KimNo ratings yet
- Assignment No 1Document5 pagesAssignment No 1Millicent LanzuelaNo ratings yet
- 305 Lab Report 1Document5 pages305 Lab Report 1munasib.ridhwaNo ratings yet
- Invertase Lab 1 and 2Document8 pagesInvertase Lab 1 and 2Tiyah TimothyNo ratings yet
- Invertase Lab 1 or 2Document8 pagesInvertase Lab 1 or 2Tiyah TimothyNo ratings yet
- Molecular Biology Lab Genomic DNA ExtractionDocument19 pagesMolecular Biology Lab Genomic DNA ExtractionVineet Kumar ThakurNo ratings yet
- Genetics Midterm 2 Study Guide PT 2 Molecular GeneticsDocument6 pagesGenetics Midterm 2 Study Guide PT 2 Molecular GeneticsCheyenne MartinsNo ratings yet
- Molecular Biology - Amity University RajasthanDocument13 pagesMolecular Biology - Amity University Rajasthanabash_u1No ratings yet
- Gel Electrophoresis of ProteinsFrom EverandGel Electrophoresis of ProteinsMichael J DunnNo ratings yet
- Stable Mutated Tau441 Transfected SH-SY5Y Cells As Screening Tool For Alzheimer 'S Disease Drug CandidatesDocument12 pagesStable Mutated Tau441 Transfected SH-SY5Y Cells As Screening Tool For Alzheimer 'S Disease Drug CandidatesShreeya BhatNo ratings yet
- Vector AppDocument5 pagesVector AppShreeya BhatNo ratings yet
- Action Plan Shreya BhatDocument5 pagesAction Plan Shreya BhatShreeya BhatNo ratings yet
- Bio-Signaling Jshreyabhat PDFDocument18 pagesBio-Signaling Jshreyabhat PDFShreeya BhatNo ratings yet
- 2ba18bt017 Shreya PDFDocument8 pages2ba18bt017 Shreya PDFShreeya BhatNo ratings yet
- 2ba18bt017 ShreyaDocument8 pages2ba18bt017 ShreyaShreeya BhatNo ratings yet
- A Beginner's Guide To Bioprocess Modes - Batch, Fed-Batch, and Continuous FermentationDocument16 pagesA Beginner's Guide To Bioprocess Modes - Batch, Fed-Batch, and Continuous FermentationShreeya BhatNo ratings yet
- ImmunoDocument7 pagesImmunoShreeya BhatNo ratings yet
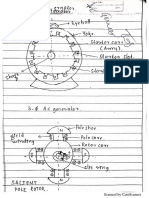
- Ac GeneratorDocument12 pagesAc GeneratorShreeya BhatNo ratings yet
- ImmunodeficiencyDocument2 pagesImmunodeficiencyShreeya BhatNo ratings yet
- Biostatistics Lab - 4 SemDocument17 pagesBiostatistics Lab - 4 SemShreeya BhatNo ratings yet
- Structural Biology AssignmentDocument8 pagesStructural Biology AssignmentShreeya BhatNo ratings yet
- Digital Communication: 1 InstructionsDocument2 pagesDigital Communication: 1 InstructionsShreeya BhatNo ratings yet
- GavanaDocument46 pagesGavanaShreeya BhatNo ratings yet
- The Fitness Project 2018 PDFDocument34 pagesThe Fitness Project 2018 PDFShreeya BhatNo ratings yet
- Restriction Digestion of Lambda DNA: InstructionDocument5 pagesRestriction Digestion of Lambda DNA: InstructionShreeya BhatNo ratings yet
- Results To Be Announced Later Basaveshwar Engineering College (Autonomous), Bagalkot - 587 103Document15 pagesResults To Be Announced Later Basaveshwar Engineering College (Autonomous), Bagalkot - 587 103Shreeya BhatNo ratings yet
- EXPERT-JSA Structural, Piping & Welding WorksDocument9 pagesEXPERT-JSA Structural, Piping & Welding Worksarun vijayNo ratings yet
- Rubrics CE ORIENT Written ReportDocument2 pagesRubrics CE ORIENT Written ReportjocelNo ratings yet
- AJPDocument35 pagesAJPAnonymous g5Xzu4V8No ratings yet
- Daftar - Pd-Upt SMP Islam Terpadu Wihdatul Ummah-2023!09!26 14-58-25Document84 pagesDaftar - Pd-Upt SMP Islam Terpadu Wihdatul Ummah-2023!09!26 14-58-25Nursyahriani SariNo ratings yet
- AdmitCard PDFDocument2 pagesAdmitCard PDFSonu Kumar KumarNo ratings yet
- Midterm Examination 1aDocument5 pagesMidterm Examination 1aEdson Louis ParaNo ratings yet
- Road Transportation System As A Viable Tool For Economic Development in Nigeria.Document12 pagesRoad Transportation System As A Viable Tool For Economic Development in Nigeria.opata cletusNo ratings yet
- Indian Companies ActDocument51 pagesIndian Companies ActvaibhavNo ratings yet
- Ijesit201601 23 PDFDocument10 pagesIjesit201601 23 PDFMouaad BousounaNo ratings yet
- Earthquake Drill Evaluation FormDocument1 pageEarthquake Drill Evaluation FormAdrin MejiaNo ratings yet
- Sales and Marketing PlanDocument10 pagesSales and Marketing PlanHuong Tran NgocNo ratings yet
- Grade 9Document60 pagesGrade 9Самал АжбеноваNo ratings yet
- QM-001 QMS-Quality ManualDocument13 pagesQM-001 QMS-Quality ManualFERNANDO MORANTESNo ratings yet
- Zoya Parasher - 2152916 - Big DataDocument6 pagesZoya Parasher - 2152916 - Big DataZoya ParasherNo ratings yet
- Sociolinguistic Survey of Northern Pakistan Volume 4Document194 pagesSociolinguistic Survey of Northern Pakistan Volume 4bashoooo1No ratings yet
- Michael Johnson Plea AgreementDocument15 pagesMichael Johnson Plea AgreementTheCullmanTribuneNo ratings yet
- SWOT AnalysisDocument2 pagesSWOT AnalysisRudford GectoNo ratings yet
- Brigandine Equipment IDs ListDocument9 pagesBrigandine Equipment IDs ListRekz ToNo ratings yet
- Presentation Dignity Slides FinalDocument105 pagesPresentation Dignity Slides FinalShae AmoresNo ratings yet
- Dolphin Station - Pre App Letter of IntentDocument3 pagesDolphin Station - Pre App Letter of Intentthe next miamiNo ratings yet
- DocuCentre S2520 S2320Document4 pagesDocuCentre S2520 S2320nuwari fadliNo ratings yet
- Building Social TiesDocument48 pagesBuilding Social TiesGissele AbolucionNo ratings yet
- SWOT Analysis of Coca Cola: StrengthsDocument6 pagesSWOT Analysis of Coca Cola: StrengthsFaisal LukyNo ratings yet
- Science Technology and Society MidtermsDocument16 pagesScience Technology and Society MidtermsDARYL JAMES MISANo ratings yet
- ChallengesDocument5 pagesChallengesMERVE İREM ALTUNTAŞNo ratings yet