You might also like
- Catalog VALSIR Canalizare PDFDocument60 pagesCatalog VALSIR Canalizare PDFAlexandru Ilioiu100% (1)
- Final Year Project Edi IrawanDocument75 pagesFinal Year Project Edi IrawanEdi IrawanNo ratings yet
- DSP UPG CU EngDocument1 pageDSP UPG CU EngArtur KwiatkowskiNo ratings yet
- Karakteristik PPDocument12 pagesKarakteristik PPRestu KusumawardaniNo ratings yet
- INTRODUCTION TO FOOD PACKAGING Chapter 5Document21 pagesINTRODUCTION TO FOOD PACKAGING Chapter 5Suleyman MiftahNo ratings yet
- Biodegradable Polymer Blends Based On Thermoplastic StarchDocument17 pagesBiodegradable Polymer Blends Based On Thermoplastic Starchelias llamasNo ratings yet
- Polymers 14 01626 v3Document44 pagesPolymers 14 01626 v3Pablo RaimondaNo ratings yet
- Distilasi PDFDocument8 pagesDistilasi PDFLusiana Eka KurniawatiNo ratings yet
- Hindaw I Publishing Cor P or Ation in Ter N Ational Jour N Al of Po Ly Mer S Cience Vo Lume 2012, Ar Ticle I D 302029, 11 Pages Doi:10.1155/2012/302029Document12 pagesHindaw I Publishing Cor P or Ation in Ter N Ational Jour N Al of Po Ly Mer S Cience Vo Lume 2012, Ar Ticle I D 302029, 11 Pages Doi:10.1155/2012/302029Sheren Mohamed NagibNo ratings yet
- Making Plastics From Monomer To PolymerDocument6 pagesMaking Plastics From Monomer To PolymerMichell Camargo JimenezNo ratings yet
- Migration of Phthalate and Non PhthalateDocument14 pagesMigration of Phthalate and Non PhthalateFREDY CORREANo ratings yet
- Biological Degradation of Polymers in The Environment: John A. GlaserDocument22 pagesBiological Degradation of Polymers in The Environment: John A. GlaserRahaf AljboriNo ratings yet
- Hypercrosslinked Polymers A ReviewDocument42 pagesHypercrosslinked Polymers A ReviewnkazemiNo ratings yet
- High Barrier Biodegradable Food PackagingDocument5 pagesHigh Barrier Biodegradable Food PackagingSarfrazNo ratings yet
- Advanced Industrial and Engineering Polymer Research: Manas ChandaDocument18 pagesAdvanced Industrial and Engineering Polymer Research: Manas ChandaElena RomeroNo ratings yet
- Effect of Chlorine Dioxide GasDocument9 pagesEffect of Chlorine Dioxide GasCarinna Saldaña - PierardNo ratings yet
- Water Vapor Transport Properties of Polyurethane FDocument13 pagesWater Vapor Transport Properties of Polyurethane FRosmery Naupari AlvarezNo ratings yet
- Mechanical Color and Barrier Properties of Biodegradable Nanocomposites Polylactic Acidnanoclay 2155 6199 1000455Document5 pagesMechanical Color and Barrier Properties of Biodegradable Nanocomposites Polylactic Acidnanoclay 2155 6199 1000455yurinnelNo ratings yet
- Extrusion Blow MoldingDocument12 pagesExtrusion Blow MoldingmaheshgupteNo ratings yet
- Mechanical Properties Molecular Structures HDPE Polymers Accepted-ManuscriptDocument29 pagesMechanical Properties Molecular Structures HDPE Polymers Accepted-Manuscriptsiddiqui4303379No ratings yet
- Polymer Engineering Sci - 2022 - Ramezani Dana - Synthesis Properties and Applications of Polylactic Acid BasedDocument22 pagesPolymer Engineering Sci - 2022 - Ramezani Dana - Synthesis Properties and Applications of Polylactic Acid Basedsabitapurohit04No ratings yet
- Study of The Degradation Mechanisms of Polyethylene During ReprocessingDocument9 pagesStudy of The Degradation Mechanisms of Polyethylene During ReprocessingJoselyn GaliciaNo ratings yet
- A Review On The Thermomechanical Properties and Biodegradation BehaviourDocument31 pagesA Review On The Thermomechanical Properties and Biodegradation BehaviourvalentinaNo ratings yet
- Hydrolytic Degradation of Bio-Based Polyesters-Effect of PH and TimeDocument8 pagesHydrolytic Degradation of Bio-Based Polyesters-Effect of PH and TimeAnnie LauNo ratings yet
- Qasim 2019 - Soil Burial Degradation of Polypropylene Starch BlendDocument7 pagesQasim 2019 - Soil Burial Degradation of Polypropylene Starch BlendDibe DivolorNo ratings yet
- Adv Ind & Eng Polymer Research 5 (2022) 60-69 - Chapter 2 - High-Performance Fibers and Tapes Based On PP and PEDocument10 pagesAdv Ind & Eng Polymer Research 5 (2022) 60-69 - Chapter 2 - High-Performance Fibers and Tapes Based On PP and PEAlexandre Ferro RochaNo ratings yet
- Progress in Organic Coatings: Short CommunicationDocument5 pagesProgress in Organic Coatings: Short CommunicationJuan Manuel Montoya SalazarNo ratings yet
- 2018 JMEMS AdhesionDocument12 pages2018 JMEMS AdhesionDr. Alois FabianiNo ratings yet
- CCL 31 2 365Document4 pagesCCL 31 2 365Fahtin NabilahNo ratings yet
- European Polymer Journal: Marina P. Arrieta, Juan López, Alberto Hernández, Emilio RayónDocument16 pagesEuropean Polymer Journal: Marina P. Arrieta, Juan López, Alberto Hernández, Emilio RayónORGEX TMNo ratings yet
- 0424 Ecsmge 2019 - VerstDocument9 pages0424 Ecsmge 2019 - VerstVetriselvan ArumugamNo ratings yet
- Polymers 13 01309Document19 pagesPolymers 13 01309Huy Tuan QuachNo ratings yet
- IPTC 11465 Enhancing Natural Gas Purification With Advanced Polymer/Molecular Sieve Composite MembranesDocument6 pagesIPTC 11465 Enhancing Natural Gas Purification With Advanced Polymer/Molecular Sieve Composite MembranesKaroll GeraldineNo ratings yet
- Io PlasticsDocument7 pagesIo PlasticserpandianNo ratings yet
- Valsir C PP l02882Document60 pagesValsir C PP l02882Budihardjo Sarwo SastrosudiroNo ratings yet
- Surfacebulk PDFDocument11 pagesSurfacebulk PDFVishwarup GoswamiNo ratings yet
- TrendsMbDesignPervaporation Subm PDFDocument28 pagesTrendsMbDesignPervaporation Subm PDFLugon HuaccachiNo ratings yet
- Cheng2021 Article OxygenBarrierFilmsOfScCO2-assiDocument16 pagesCheng2021 Article OxygenBarrierFilmsOfScCO2-assiPablo RaimondaNo ratings yet
- Processing and CharacterizationDocument18 pagesProcessing and CharacterizationKawaiiBunnehSukiNo ratings yet
- Muthu Raj 2015Document13 pagesMuthu Raj 2015Shivani BehareNo ratings yet
- Characterization of Metallized Biaxially Oriented Polypropylene FilmDocument10 pagesCharacterization of Metallized Biaxially Oriented Polypropylene FilmLaboratory Plant 7No ratings yet
- NCL Report - Chapter 4Document6 pagesNCL Report - Chapter 4Aparna YaduNo ratings yet
- The Effect of Extensive Mechanical RecycDocument11 pagesThe Effect of Extensive Mechanical RecycAhmet AltunNo ratings yet
- HdpeDocument9 pagesHdpeXuân Giang NguyễnNo ratings yet
- Permeation - Oxygen Packaging - 18 - 2016Document11 pagesPermeation - Oxygen Packaging - 18 - 2016muriloinnocentiniNo ratings yet
- Activation Energy For The Pyrolysis of Polymer WastesDocument6 pagesActivation Energy For The Pyrolysis of Polymer WastesSwiftTGSolutionsNo ratings yet
- Hidrolisis, Pirosis....Document26 pagesHidrolisis, Pirosis....Elena RomeroNo ratings yet
- Advances in Polyamide Nanocomposites A ReviewDocument20 pagesAdvances in Polyamide Nanocomposites A ReviewSalman khanNo ratings yet
- Polymers 14 02317Document35 pagesPolymers 14 02317Ana ursNo ratings yet
- Evaluation and Identification of Degradative Processes in Post Consumer Recycled HDPEDocument7 pagesEvaluation and Identification of Degradative Processes in Post Consumer Recycled HDPEAroop Ratan SenNo ratings yet
- Resources, Conservation & Recycling: ReviewDocument25 pagesResources, Conservation & Recycling: ReviewElena RomeroNo ratings yet
- App 38945Document11 pagesApp 38945Annisya ZahraNo ratings yet
- Supercritical Gel Drying: A Powerful Tool For Tailoring Symmetric Porous PVDF-HFP MembranesDocument10 pagesSupercritical Gel Drying: A Powerful Tool For Tailoring Symmetric Porous PVDF-HFP MembranesArdila Hayu TiwikramaNo ratings yet
- Lecture FIVE: Classification of Polymer MaterialsDocument8 pagesLecture FIVE: Classification of Polymer MaterialsSadiq SalamNo ratings yet
- Mechanical, Color and Barrier, Properties of Biodegradable. 2018Document7 pagesMechanical, Color and Barrier, Properties of Biodegradable. 2018yurinnelNo ratings yet
- Polymers and Polymers BlendsDocument51 pagesPolymers and Polymers Blendsziradagreat539No ratings yet
- Study of Liquid CrystalDocument10 pagesStudy of Liquid CrystalXinshi ChenNo ratings yet
- 1 s2.0 S0379677997806583 Main PDFDocument8 pages1 s2.0 S0379677997806583 Main PDFMuhammad Faisal AminNo ratings yet
- Articulo 6 PDFDocument7 pagesArticulo 6 PDFDeiby LealNo ratings yet
- Plastic Polymers PDFDocument8 pagesPlastic Polymers PDFnoor emadNo ratings yet
- Atmospheric Pressure Plasma Treatment of Polymers: Relevance to AdhesionFrom EverandAtmospheric Pressure Plasma Treatment of Polymers: Relevance to AdhesionNo ratings yet
- Plastics as Corrosion-Resistant Materials: The Commonwealth and International Library: Plastics DivisionFrom EverandPlastics as Corrosion-Resistant Materials: The Commonwealth and International Library: Plastics DivisionNo ratings yet
- Current Research Trends On Plastic Pollution and Ecological Impacts OnDocument9 pagesCurrent Research Trends On Plastic Pollution and Ecological Impacts OnMaria-MirabelaGherasimNo ratings yet
- Beyond Mechanical Recycling Giving New Life To Plastic WasteDocument38 pagesBeyond Mechanical Recycling Giving New Life To Plastic WasteChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Solutions and Integrated Strategies For The Control and Mitigation of Plastic and Microplastic PollutionDocument19 pagesSolutions and Integrated Strategies For The Control and Mitigation of Plastic and Microplastic Pollutionashadi asriNo ratings yet
- Solvent-Based Separation and Recycling of Waste Plastics A ReviewDocument14 pagesSolvent-Based Separation and Recycling of Waste Plastics A ReviewChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Sub - and Supercritical Water For Chemical Recycling of Plastic WasteDocument22 pagesSub - and Supercritical Water For Chemical Recycling of Plastic WasteChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Shah2008 PDFDocument20 pagesShah2008 PDFadri2115No ratings yet
- Increased Plastic Pollution Due To COVID-19 Pandemic Challenges andDocument9 pagesIncreased Plastic Pollution Due To COVID-19 Pandemic Challenges andChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Biodegradation of Micro-Polyethylene Particles by BacterialDocument7 pagesBiodegradation of Micro-Polyethylene Particles by BacterialChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Polymer-Plastics Technology and EngineeringDocument9 pagesPolymer-Plastics Technology and EngineeringChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastics MaterialsDocument206 pagesPlastics MaterialsChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Recently Emerging Trends in PolymerDocument57 pagesRecently Emerging Trends in PolymerChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastic Waste As A Global Challengeare Biodegradable PlasticsDocument9 pagesPlastic Waste As A Global Challengeare Biodegradable PlasticsChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastics Recycling Challenges and OpportunitiesDocument13 pagesPlastics Recycling Challenges and OpportunitiesChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Sub - and Supercritical Water For Chemical Recycling of Plastic WasteDocument22 pagesSub - and Supercritical Water For Chemical Recycling of Plastic WasteChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastic Pollutantseffective Waste Management For PollutionDocument13 pagesPlastic Pollutantseffective Waste Management For PollutionChristhy Vanessa Ruiz MadroñeroNo ratings yet
- A Life Cycle Assessment ofDocument18 pagesA Life Cycle Assessment ofChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Methods For Plastic RecyclingDocument19 pagesMethods For Plastic RecyclingAshik Shah100% (2)
- Recycling of PET: Firas Awaja, Dumitru PavelDocument25 pagesRecycling of PET: Firas Awaja, Dumitru PavelFernanda TenesacaNo ratings yet
- Plastics Recycling Challenges and OpportunitiesDocument13 pagesPlastics Recycling Challenges and OpportunitiesChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastics Recycling Challenges and OpportunitiesDocument13 pagesPlastics Recycling Challenges and OpportunitiesChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Polymer-Plastics Technology and EngineeringDocument9 pagesPolymer-Plastics Technology and EngineeringChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Plastic Pollutantseffective Waste Management For PollutionDocument13 pagesPlastic Pollutantseffective Waste Management For PollutionChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Recovery of PolyhydroxyalkanoatesDocument45 pagesRecovery of PolyhydroxyalkanoatesChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Dehydration of Waterplasticized Poly (Vinylalcohol) Systemsparticular Behavior Ofisothermal Mass TransferDocument8 pagesDehydration of Waterplasticized Poly (Vinylalcohol) Systemsparticular Behavior Ofisothermal Mass TransferChristhy Vanessa Ruiz MadroñeroNo ratings yet
- IntercalationCarboxylicAcids LDHDocument12 pagesIntercalationCarboxylicAcids LDHChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Characterization of Polyvinyl AlcoholDocument11 pagesCharacterization of Polyvinyl AlcoholChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Polymer-Plastics Technology and EngineeringDocument9 pagesPolymer-Plastics Technology and EngineeringChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Recently Emerging Trends in PolymerDocument57 pagesRecently Emerging Trends in PolymerChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Crystal Structure of PVADocument2 pagesCrystal Structure of PVAChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Research ProposalDocument2 pagesResearch Proposalsmh9662No ratings yet
- Generation Transmission and Distribution Notes.Document88 pagesGeneration Transmission and Distribution Notes.Abhishek NirmalkarNo ratings yet
- BLE Catalogue 2013Document21 pagesBLE Catalogue 2013Shahina Parvin ShaikNo ratings yet
- Physics 715 HW 1Document13 pagesPhysics 715 HW 1Antonildo PereiraNo ratings yet
- BarDocument1 pageBarJoannalyn Libo-onNo ratings yet
- كتالوج وصفي للعملات الرومانية النادرة وغير المحررة من الفترة الأولى للعملات المعدنية الرومانية إلى انقراض الإمبراطورية تحت حكDocument570 pagesكتالوج وصفي للعملات الرومانية النادرة وغير المحررة من الفترة الأولى للعملات المعدنية الرومانية إلى انقراض الإمبراطورية تحت حكFayza MaghrabiNo ratings yet
- 990XP Bandit ChipperDocument5 pages990XP Bandit ChipperFrancisco ConchaNo ratings yet
- Brand Loyalty & Competitive Analysis of Pankaj NamkeenDocument59 pagesBrand Loyalty & Competitive Analysis of Pankaj NamkeenBipin Bansal Agarwal100% (1)
- Cognitive Benefits of Language LearningDocument11 pagesCognitive Benefits of Language LearningIlhamdi HafizNo ratings yet
- CREDEDocument10 pagesCREDEDaffodilsNo ratings yet
- Teacher Thought For InterviewDocument37 pagesTeacher Thought For InterviewMahaprasad JenaNo ratings yet
- Anti LeproticDocument9 pagesAnti LeproticMeenakshi shARMANo ratings yet
- PlayStation MagazineDocument116 pagesPlayStation MagazineFrank Costello67% (3)
- Exercise 3 - Wireframe Geometry Creation and Editing - Rev ADocument33 pagesExercise 3 - Wireframe Geometry Creation and Editing - Rev AdevNo ratings yet
- John Deere CaseDocument2 pagesJohn Deere CaseAldo ReynaNo ratings yet
- Screen 2014 Uricchio 119 27Document9 pagesScreen 2014 Uricchio 119 27NazishTazeemNo ratings yet
- BS en 13369-2018 - TC - (2020-11-30 - 09-45-34 Am)Document164 pagesBS en 13369-2018 - TC - (2020-11-30 - 09-45-34 Am)Mustafa Uzyardoğan100% (1)
- Advantage Dis OqpskDocument5 pagesAdvantage Dis OqpskHarun AminurasyidNo ratings yet
- RTI SpicesDocument226 pagesRTI SpicesvivebajajNo ratings yet
- Q1 WK 2 To 3 Las Fabm2 Kate DionisioDocument8 pagesQ1 WK 2 To 3 Las Fabm2 Kate DionisioFunji BuhatNo ratings yet
- Language - Introduction To The Integrated Language Arts CompetenciesDocument7 pagesLanguage - Introduction To The Integrated Language Arts CompetenciesHari Ng Sablay100% (1)
- With Pneumatic and Electric Actuators: Datasheet 448001 EnglishDocument7 pagesWith Pneumatic and Electric Actuators: Datasheet 448001 EnglishPinak ProjectsNo ratings yet
- eLearnMarkets OptionsBuying HindiDocument17 pageseLearnMarkets OptionsBuying Hindisrinivas20% (1)
- FreeBSD HandbookDocument26 pagesFreeBSD Handbookhembeck119No ratings yet
- Cement Grouted Rock BoltsDocument28 pagesCement Grouted Rock BoltsBhaskar ReddyNo ratings yet
- Analysis and Design of Suspended Buildings: Creative and Innovative Report-1Document14 pagesAnalysis and Design of Suspended Buildings: Creative and Innovative Report-1Nirmal RaviNo ratings yet
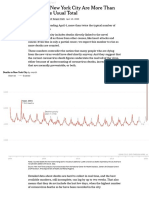
- Deaths in New York City Are More Than Double The Usual TotalDocument3 pagesDeaths in New York City Are More Than Double The Usual TotalRamón RuizNo ratings yet
- Draft Technical Notes SGLG 2023 - National OrientationDocument109 pagesDraft Technical Notes SGLG 2023 - National OrientationZane ZyneNo ratings yet