You might also like
- Carrying Out Gas-Liquid ChromatographyDocument9 pagesCarrying Out Gas-Liquid ChromatographymahtabsirNo ratings yet
- Gas Liquid ChromatographyDocument8 pagesGas Liquid Chromatographyindra_f04No ratings yet
- How The Column WorksDocument5 pagesHow The Column Worksalex_andra_22No ratings yet
- Working Principle of GCDocument4 pagesWorking Principle of GCAmnaKamranNo ratings yet
- Analaytical Assignment Group-3Document10 pagesAnalaytical Assignment Group-3yilma wendayehuNo ratings yet
- Melting PointDocument4 pagesMelting PointShama Al-ShadidiNo ratings yet
- Summited By: - Submitted To: - Riteish Sharma Mrs. Chhavi SharmaDocument23 pagesSummited By: - Submitted To: - Riteish Sharma Mrs. Chhavi Sharmapyiush jainNo ratings yet
- Year 10 Book 2015Document188 pagesYear 10 Book 2015api-317749980No ratings yet
- Liquid-Liquid Chromatography ExplainedDocument5 pagesLiquid-Liquid Chromatography ExplainedAlaeldeen EltayebNo ratings yet
- Rahul 4Document8 pagesRahul 4benkay1001No ratings yet
- Chapter 22 - Gas ChromatographyDocument12 pagesChapter 22 - Gas ChromatographyJanani AnbalaganNo ratings yet
- GC PDFDocument27 pagesGC PDFViper ThroughNo ratings yet
- Fe 205 Labaratory Report 1Document6 pagesFe 205 Labaratory Report 1Hüsamettin KirazNo ratings yet
- Research Paper On Gas ChromatographyDocument6 pagesResearch Paper On Gas Chromatographyc9rbzcr0100% (1)
- Theory RachitDocument13 pagesTheory RachitRACHITNo ratings yet
- Gas Chromatography TheoryDocument3 pagesGas Chromatography TheorymechneeNo ratings yet
- Gas ChromatographyDocument4 pagesGas ChromatographyAgustin A.No ratings yet
- Chromatography Techniques ExplainedDocument9 pagesChromatography Techniques ExplainedDeepak PradhanNo ratings yet
- Rate of Evaporation of Different Liquids-RagavDocument13 pagesRate of Evaporation of Different Liquids-RagavVijaya Ragav SNo ratings yet
- Chromatography Techniques GuideDocument19 pagesChromatography Techniques Guidewama ojhaNo ratings yet
- Theory: Factors Influencing Rate of EvaporationDocument10 pagesTheory: Factors Influencing Rate of Evaporationviraj muzumdarNo ratings yet
- Natural Gas EngineeringDocument6 pagesNatural Gas EngineeringAnonymous id2bXjUJNo ratings yet
- Experiments on Evaporation RatesDocument12 pagesExperiments on Evaporation RatesArkodeep PodderNo ratings yet
- HPLC Guide: Separate Mixtures With High-Performance Liquid ChromatographyDocument8 pagesHPLC Guide: Separate Mixtures With High-Performance Liquid ChromatographyDeepanshi BansalNo ratings yet
- 45 GC PDFDocument2 pages45 GC PDFFaizal ThelordImmortalNo ratings yet
- ReportDocument7 pagesReportShōyōHinataNo ratings yet
- Determination of Rate of Evaporation of Different Liquids.Document22 pagesDetermination of Rate of Evaporation of Different Liquids.Jinesh Doshi33% (6)
- Rate of Evaporation of Different LiquidsDocument5 pagesRate of Evaporation of Different LiquidsMayankJainNo ratings yet
- Chemisty Igcse Updated Till SyllabusDocument97 pagesChemisty Igcse Updated Till Syllabusapi-181176018No ratings yet
- Melting Point: Point at Which Solid and Liquid Phases Are in EquilibriumDocument6 pagesMelting Point: Point at Which Solid and Liquid Phases Are in EquilibriumPhú NguyễnNo ratings yet
- Experiment 6 - DistillationDocument5 pagesExperiment 6 - DistillationRohit BiswasNo ratings yet
- Group Members: Suhaidah Binti Nazori UK21425 Mohd Faizuddin Bin Abu Hasan UK21455 Siti Fasihah Binti Rameli UK21478Document27 pagesGroup Members: Suhaidah Binti Nazori UK21425 Mohd Faizuddin Bin Abu Hasan UK21455 Siti Fasihah Binti Rameli UK21478ah_16036566No ratings yet
- Understanding the States of Matter and Particle TheoryDocument10 pagesUnderstanding the States of Matter and Particle TheoryAjitabh SinghNo ratings yet
- Gas ChromatographyDocument80 pagesGas ChromatographyNeham Baroha100% (2)
- BIO 105L - CHAPTER 6 - Melting Point DeteminationDocument4 pagesBIO 105L - CHAPTER 6 - Melting Point DeteminationFranchiezca AoananNo ratings yet
- Board ProjectpdfDocument30 pagesBoard ProjectpdfN2K FF GamingNo ratings yet
- GC Column Efficiency and Band BroadeningDocument25 pagesGC Column Efficiency and Band BroadeningPrabneeshNo ratings yet
- Study of Rate of Evaporation of LiquidsDocument18 pagesStudy of Rate of Evaporation of Liquidsrohinrohin78% (9)
- CHM170L Exp1 Determination of Molar MassDocument5 pagesCHM170L Exp1 Determination of Molar MassKaiser SaltoNo ratings yet
- Rate of Evaporation FactorsDocument9 pagesRate of Evaporation FactorsRishabhNo ratings yet
- Gas ChromatographyDocument12 pagesGas ChromatographyYaman Adnan QabajaNo ratings yet
- Vacuum PumpDocument4 pagesVacuum PumpJohnNo ratings yet
- Adarsh Kumar Chemistry ProjectDocument15 pagesAdarsh Kumar Chemistry Projectletsgoiit343No ratings yet
- HPLC Report2019Document11 pagesHPLC Report2019Nancy SrivastavaNo ratings yet
- Gas ChromatographyDocument5 pagesGas ChromatographyassertivailNo ratings yet
- High Performance Liquid ChromatographyDocument4 pagesHigh Performance Liquid ChromatographyRavin KumarNo ratings yet
- Igcse Complete Chemistry Notes: Unit 1: States of MatterDocument72 pagesIgcse Complete Chemistry Notes: Unit 1: States of MatterYoga RomdoniNo ratings yet
- Lab Report-11: Environmental Chemistry (ENE-213) Course Instructor: Dr. Sofia BaigDocument7 pagesLab Report-11: Environmental Chemistry (ENE-213) Course Instructor: Dr. Sofia BaigHaniya SiddiqueNo ratings yet
- A ChromglcDocument2 pagesA ChromglcWilliam TsuiNo ratings yet
- AasDocument11 pagesAasLovely Pearl Reyes ValendezNo ratings yet
- Unit-3Document25 pagesUnit-3sankar velisettyNo ratings yet
- Chem Investigatory ProjectDocument10 pagesChem Investigatory ProjectSS RUTVIJ REDDYNo ratings yet
- ChromatographyDocument8 pagesChromatographyThangavel SarujanNo ratings yet
- Molar Mass of A Volatile LiquidDocument16 pagesMolar Mass of A Volatile LiquidJoaquin MoulicNo ratings yet
- A Partial Physics BreakdownDocument13 pagesA Partial Physics BreakdownKamochiUzumakiNo ratings yet
- A System of Instruction in the Practical Use of the Blowpipe Being A Graduated Course Of Analysis For The Use Of Students And All Those Engaged In The Examination Of Metallic CombinationsFrom EverandA System of Instruction in the Practical Use of the Blowpipe Being A Graduated Course Of Analysis For The Use Of Students And All Those Engaged In The Examination Of Metallic CombinationsNo ratings yet
- Physico-Chemistry of Solid-Gas Interfaces: Concepts and Methodology for Gas Sensor DevelopmentFrom EverandPhysico-Chemistry of Solid-Gas Interfaces: Concepts and Methodology for Gas Sensor DevelopmentNo ratings yet
- A System of Instruction in the Practical Use of the BlowpipeFrom EverandA System of Instruction in the Practical Use of the BlowpipeNo ratings yet
- Wer 1408Document7 pagesWer 1408Faisal MumtazNo ratings yet
- Summary of A Conceptual Design of Low Fouling and High Recovery FO-MSF-DESKTOP-47LO0PODocument1 pageSummary of A Conceptual Design of Low Fouling and High Recovery FO-MSF-DESKTOP-47LO0POFaisal MumtazNo ratings yet
- BQTS - WetWellCalcDocument3 pagesBQTS - WetWellCalcFaisal MumtazNo ratings yet
- EENG703 - Assignment04 (AutoRecovered)Document19 pagesEENG703 - Assignment04 (AutoRecovered)Faisal MumtazNo ratings yet
- Optimisation of The WWTP Performance MakiniaDocument39 pagesOptimisation of The WWTP Performance MakiniaFaisal MumtazNo ratings yet
- Hydraulic SizingDocument2 pagesHydraulic SizingFaisal MumtazNo ratings yet
- Summary of Development of A Steady-State Mathematical Model For MEE-TVCDocument2 pagesSummary of Development of A Steady-State Mathematical Model For MEE-TVCFaisal MumtazNo ratings yet
- Calculation of Water Flow Rates For Different Pipe Sizes - US UnitsDocument4 pagesCalculation of Water Flow Rates For Different Pipe Sizes - US UnitsRyan KaneNo ratings yet
- ExergyDocument10 pagesExergyFaisal MumtazNo ratings yet
- Mannings MetricDocument1 pageMannings MetricFaisal MumtazNo ratings yet
- Summary of Vector and Tensor NotationDocument41 pagesSummary of Vector and Tensor NotationFaisal MumtazNo ratings yet
- Friction FactorDocument6 pagesFriction Factorrajeshsapkota123No ratings yet
- IWTP8-MFQ-ILF-21-0500-Mechanical Equipment List 23-p2mDocument10 pagesIWTP8-MFQ-ILF-21-0500-Mechanical Equipment List 23-p2mFaisal MumtazNo ratings yet
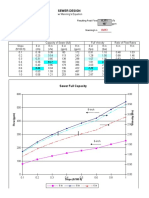
- WWSewerDesign (EDocFind - Com) - 1Document10 pagesWWSewerDesign (EDocFind - Com) - 1Saw ThreedNo ratings yet
- 05 - Design of Package RCTRDocument1 page05 - Design of Package RCTRFaisal MumtazNo ratings yet
- Notes 3: Mass TransportDocument60 pagesNotes 3: Mass TransportFaisal MumtazNo ratings yet
- Sizing PoleniDocument87 pagesSizing PoleniFaisal MumtazNo ratings yet
- 06 - STP Energy Balance (Power Consumption Calcs)Document7 pages06 - STP Energy Balance (Power Consumption Calcs)Faisal MumtazNo ratings yet
- Mass and Energy Balances of Sludge Processing in Reference and Upgraded Waste Water Treatment PlantsDocument14 pagesMass and Energy Balances of Sludge Processing in Reference and Upgraded Waste Water Treatment Plantsmohit guptaNo ratings yet
- Assignment 1Document3 pagesAssignment 1Faisal MumtazNo ratings yet
- 00 - WWT EngineeringDocument188 pages00 - WWT EngineeringFaisal MumtazNo ratings yet
- 02 - Case Study - Industrial Technical QueryDocument3 pages02 - Case Study - Industrial Technical QueryFaisal MumtazNo ratings yet
- Experimental Study of Thermal and Catalytic Pyrolysis of Plastic Waste ComponentsDocument11 pagesExperimental Study of Thermal and Catalytic Pyrolysis of Plastic Waste ComponentsFaisal MumtazNo ratings yet
- RSC Advances: PaperDocument14 pagesRSC Advances: PaperFaisal MumtazNo ratings yet
- Pyrolysis of Polypropylene Waste With Natural Zeolite As CatalystDocument10 pagesPyrolysis of Polypropylene Waste With Natural Zeolite As CatalystFaisal MumtazNo ratings yet
- Isoconversional Methods - Fundamentals, Meaning and ApplicationDocument11 pagesIsoconversional Methods - Fundamentals, Meaning and ApplicationFaisal MumtazNo ratings yet
- 01 - Mass & Energy Balance WWT (CHP3)Document56 pages01 - Mass & Energy Balance WWT (CHP3)Faisal MumtazNo ratings yet
- A Study On Thermo - Catalytic Degradation of PET (Polyethylene Terephthalate) Waste For Fuel Production and Chemical ProductsDocument6 pagesA Study On Thermo - Catalytic Degradation of PET (Polyethylene Terephthalate) Waste For Fuel Production and Chemical ProductsFaisal MumtazNo ratings yet
- PDF Created With Pdffactory Trial VersionDocument1 pagePDF Created With Pdffactory Trial VersionFaisal MumtazNo ratings yet
- PDF Created With Pdffactory Trial VersionDocument1 pagePDF Created With Pdffactory Trial VersionFaisal MumtazNo ratings yet
- 99th Indian Science Congress (Bhubaneshwar)Document95 pages99th Indian Science Congress (Bhubaneshwar)Aadarsh DasNo ratings yet
- C11 Strategy DevelopmentDocument30 pagesC11 Strategy DevelopmentPARTI KEADILAN RAKYAT NIBONG TEBALNo ratings yet
- ElectricalDocument30 pagesElectricalketerNo ratings yet
- 1 PBDocument11 pages1 PBAnggita Wulan RezkyanaNo ratings yet
- Evolution Packet FinalDocument24 pagesEvolution Packet FinalJoaquinNo ratings yet
- Multivariate Analysis Homework QuestionsDocument2 pagesMultivariate Analysis Homework Questions歐怡君No ratings yet
- An Overview of The FUPLA 2 Tools: Project DatabaseDocument2 pagesAn Overview of The FUPLA 2 Tools: Project DatabaseJulio Cesar Rojas SaavedraNo ratings yet
- 2 5 Marking ScheduleDocument6 pages2 5 Marking Scheduleapi-218511741No ratings yet
- Oracle Apps Quality ModuleDocument17 pagesOracle Apps Quality ModuleSantOsh100% (2)
- Handbook of Zen, Mindfulness and Spiritual Health PDFDocument324 pagesHandbook of Zen, Mindfulness and Spiritual Health PDFMatthew Grayson100% (3)
- SS1c MIDTERMS LearningModuleDocument82 pagesSS1c MIDTERMS LearningModuleBryce VentenillaNo ratings yet
- Vincent Ira B. Perez: Barangay Gulod, Calatagan, BatangasDocument3 pagesVincent Ira B. Perez: Barangay Gulod, Calatagan, BatangasJohn Ramsel Boter IINo ratings yet
- Friction, Gravity and Energy TransformationsDocument12 pagesFriction, Gravity and Energy TransformationsDaiserie LlanezaNo ratings yet
- ACL GRC Risk Manager - Usage Guide V1.1Document28 pagesACL GRC Risk Manager - Usage Guide V1.1Rohit ShettyNo ratings yet
- How Do I Prepare For Public Administration For IAS by Myself Without Any Coaching? Which Books Should I Follow?Document3 pagesHow Do I Prepare For Public Administration For IAS by Myself Without Any Coaching? Which Books Should I Follow?saiviswanath0990100% (1)
- Biamp Vocia Catalog Apr2020Document24 pagesBiamp Vocia Catalog Apr2020Mahavir Shantilal DhokaNo ratings yet
- One - Pager - SOGEVAC SV 320 BDocument2 pagesOne - Pager - SOGEVAC SV 320 BEOLOS COMPRESSORS LTDNo ratings yet
- NSX 100-630 User ManualDocument152 pagesNSX 100-630 User Manualagra04100% (1)
- Lateral capacity of pile in clayDocument10 pagesLateral capacity of pile in clayGeetha MaNo ratings yet
- GKInvest Market ReviewDocument66 pagesGKInvest Market ReviewjhonxracNo ratings yet
- SolarBright MaxBreeze Solar Roof Fan Brochure Web 1022Document4 pagesSolarBright MaxBreeze Solar Roof Fan Brochure Web 1022kewiso7811No ratings yet
- Plastic Welding: We Know HowDocument125 pagesPlastic Welding: We Know Howprabal rayNo ratings yet
- Cyber Security 2017Document8 pagesCyber Security 2017Anonymous i1ClcyNo ratings yet
- Masterbatch Buyers Guide PDFDocument8 pagesMasterbatch Buyers Guide PDFgurver55No ratings yet
- JTP Brochure - 2Document6 pagesJTP Brochure - 2YAKOVNo ratings yet
- CHEMICAL ANALYSIS OF WATER SAMPLEDocument5 pagesCHEMICAL ANALYSIS OF WATER SAMPLEAiron Fuentes EresNo ratings yet
- HHG4M - Lifespan Development Textbook Lesson 2Document95 pagesHHG4M - Lifespan Development Textbook Lesson 2Lubomira SucheckiNo ratings yet
- Tirfor: Lifting and Pulling Machines With Unlimited Wire RopeDocument26 pagesTirfor: Lifting and Pulling Machines With Unlimited Wire RopeGreg ArabazNo ratings yet
- Corporate Governance in SMEsDocument18 pagesCorporate Governance in SMEsSana DjaanineNo ratings yet
- Classroom Observation Form 1Document4 pagesClassroom Observation Form 1api-252809250No ratings yet