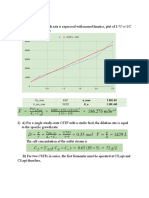
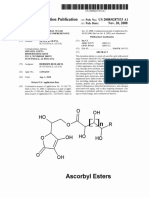
You might also like
- Astm E3-11Document12 pagesAstm E3-11Alejandro OrtizNo ratings yet
- Drugs Effects On The Central Nervous SystemDocument6 pagesDrugs Effects On The Central Nervous SystemMuhammad AriefNo ratings yet
- THERMAL PROPERTIES OF POLYMER (Update)Document38 pagesTHERMAL PROPERTIES OF POLYMER (Update)Vĩ Lê QuangNo ratings yet
- Encapsulation and Controlled ReleaseDocument29 pagesEncapsulation and Controlled ReleaseKasan BasanNo ratings yet
- REGULATORY SCIENCE OF LIPOSOME DRUGDocument6 pagesREGULATORY SCIENCE OF LIPOSOME DRUGrandatagNo ratings yet
- Enzyme Inhibition in Drug Discovery and Development: The Good and the BadFrom EverandEnzyme Inhibition in Drug Discovery and Development: The Good and the BadChuang LuNo ratings yet
- We Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsDocument51 pagesWe Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsClaudia TiffanyNo ratings yet
- 4006oral Solutions, Syrups, & ElixirsDocument83 pages4006oral Solutions, Syrups, & ElixirsOctavio Cardenas OrozcoNo ratings yet
- Quality by DesignDocument25 pagesQuality by DesignQi D'JavoeNo ratings yet
- Aerospace Material Specification: Plating, Nickel General PurposeDocument8 pagesAerospace Material Specification: Plating, Nickel General PurposeSURYAS63No ratings yet
- Polymer Microspheres For Controlled Drug Release PDFDocument18 pagesPolymer Microspheres For Controlled Drug Release PDFjmprtaNo ratings yet
- Solubility Evaluations of Osimertinib Mesylate in Physiological BuffersDocument6 pagesSolubility Evaluations of Osimertinib Mesylate in Physiological BuffersBaru Chandrasekhar RaoNo ratings yet
- Preparation of Media: Microbiology BIOL 275Document9 pagesPreparation of Media: Microbiology BIOL 275Nur AishaNo ratings yet
- Sample Problem #2Document3 pagesSample Problem #2Dozdi100% (1)
- Insilico Drug Designing for Malaria TreatmentDocument63 pagesInsilico Drug Designing for Malaria TreatmentFree Escort ServiceNo ratings yet
- Science - Stage 5 - 02 - 8RP - AFP - tcm142-640011Document16 pagesScience - Stage 5 - 02 - 8RP - AFP - tcm142-640011Abeer Elgamal100% (1)
- Phase RuleDocument43 pagesPhase RuleAltamash KhanNo ratings yet
- Ich - Introduction, Organization & GuidelinesDocument52 pagesIch - Introduction, Organization & GuidelinesALINo ratings yet
- COA PH 4 MerckDocument2 pagesCOA PH 4 MerckArdhy Lazuardy100% (1)
- Lipinski Rule of FiveDocument21 pagesLipinski Rule of FiveSasikala RajendranNo ratings yet
- Microbiology Case Study: Cryptococcal MeningitisDocument16 pagesMicrobiology Case Study: Cryptococcal MeningitisRomie SolacitoNo ratings yet
- Lipinski Rule of FiveDocument21 pagesLipinski Rule of Fivetuti ratnasari100% (1)
- 010 Ionic EquilibriaDocument16 pages010 Ionic EquilibriaStephie GhassanNo ratings yet
- Formulation and Evaluation of Darifenacin Hydrobromide Extended Release Matrix TabletsDocument32 pagesFormulation and Evaluation of Darifenacin Hydrobromide Extended Release Matrix TabletsSyed Abdul Haleem AkmalNo ratings yet
- CriptosporidiumDocument10 pagesCriptosporidiumInjektilo MendozaNo ratings yet
- Nucleus: Sudhanshu Shekhar, M.Tech BiotechDocument22 pagesNucleus: Sudhanshu Shekhar, M.Tech BiotechSudhanshu ShekharNo ratings yet
- AIDS Man Diagnosed with Cryptococcal MeningitisDocument22 pagesAIDS Man Diagnosed with Cryptococcal Meningitishus100No ratings yet
- Protecting Public Health in the Dengvaxia ControversyDocument2 pagesProtecting Public Health in the Dengvaxia ControversyJuan MatikasNo ratings yet
- Health Effects of Rancid OilsDocument3 pagesHealth Effects of Rancid OilsMaiden PEINo ratings yet
- Time Is More Valuable Than MoneyDocument2 pagesTime Is More Valuable Than Moneyjyothi vaishnavNo ratings yet
- Enzyme KineticsDocument28 pagesEnzyme KineticsJed Dumadag DanoNo ratings yet
- Lecture - 1 Dosage FormDocument13 pagesLecture - 1 Dosage FormAshique Farhad100% (1)
- Re Ections: Why Are You Afraid? Have You No Faith?Document14 pagesRe Ections: Why Are You Afraid? Have You No Faith?Zanetta JansenNo ratings yet
- En ASFA AU Koplík UV - VIS - SpectrometryDocument12 pagesEn ASFA AU Koplík UV - VIS - SpectrometryJedha YantiNo ratings yet
- CosolvancyDocument6 pagesCosolvancyyashpandya01No ratings yet
- Phase RuleDocument27 pagesPhase RulejaiminNo ratings yet
- 6 SyrupsDocument37 pages6 Syrups鄭宇揚No ratings yet
- 1 Drug Polymorphism and Dosage Form Design A Practical PerspectiveDocument13 pages1 Drug Polymorphism and Dosage Form Design A Practical Perspectivejulieth vNo ratings yet
- Properties of BiofilmDocument42 pagesProperties of BiofilmDrKrishna DasNo ratings yet
- Controlled Release Systems of BiopharmaceuticalsDocument11 pagesControlled Release Systems of BiopharmaceuticalsSabiruddin Mirza DipuNo ratings yet
- A Eutectic MixtureDocument8 pagesA Eutectic MixtureabhishekNo ratings yet
- Development of Filipino LiteratureDocument4 pagesDevelopment of Filipino LiteratureErnan Sanga MitsphNo ratings yet
- Absorption of DrugsDocument24 pagesAbsorption of DrugsShrikant SharmaNo ratings yet
- Car-T Cell Therapy - Final DraftDocument50 pagesCar-T Cell Therapy - Final DraftFatina HatoumNo ratings yet
- Physics of Tablet CompressionDocument60 pagesPhysics of Tablet CompressionSagar FirkeNo ratings yet
- Therapeutic ProteinsDocument18 pagesTherapeutic ProteinsD Mohamed R. HassanNo ratings yet
- Insulin From RecDocument6 pagesInsulin From RecDavid ZamoraNo ratings yet
- Drug Discovery Process and TargetsDocument6 pagesDrug Discovery Process and TargetsGaurav DarochNo ratings yet
- Perception of Different Sugars FinalDocument4 pagesPerception of Different Sugars FinalLourainne Faith AloceljaNo ratings yet
- Herpes Simplex Virus Type 2: Guidelines For The Management of Sexually Transmitted InfectionsDocument52 pagesHerpes Simplex Virus Type 2: Guidelines For The Management of Sexually Transmitted Infectionssgd 2No ratings yet
- Drug DesignDocument6 pagesDrug DesignMuthu LakshmiNo ratings yet
- Liposome and NanotechnologyDocument50 pagesLiposome and NanotechnologyRajesh JadonNo ratings yet
- DNA Replication Is The Basis of TheDocument42 pagesDNA Replication Is The Basis of Theapi-3800279No ratings yet
- The Role of Microorganisms in Bioremediation-A ReviewDocument9 pagesThe Role of Microorganisms in Bioremediation-A ReviewKashmeera Geethanjali AnirudhanNo ratings yet
- Session 1-1 William Potter PDFDocument21 pagesSession 1-1 William Potter PDFDrAmit Verma100% (1)
- Biopharming TrainingDocument97 pagesBiopharming TrainingBalakrishnan NatarajanNo ratings yet
- SuspensionsDocument20 pagesSuspensionsK3nsh1nXNo ratings yet
- Pharmaceutical Suspension: Nahla S. Barakat, PH.DDocument43 pagesPharmaceutical Suspension: Nahla S. Barakat, PH.DRaisa Alvina100% (1)
- Importance of Stereochemistry in Drug DevelopmentDocument44 pagesImportance of Stereochemistry in Drug DevelopmentChigozie EzeanoketeNo ratings yet
- Unit 6 Targeted Drug DeliveryDocument42 pagesUnit 6 Targeted Drug Deliveryabdullah2020100% (1)
- Non Clinical Drug DevelopmentDocument75 pagesNon Clinical Drug DevelopmentalexNo ratings yet
- In Vitro Dissolution Testing Models: DR Rajesh MujariyaDocument35 pagesIn Vitro Dissolution Testing Models: DR Rajesh MujariyaRajesh MujariyaNo ratings yet
- Organic Pharmaceutical Chemistry IV 1st Semester, Year 5 (2016-2017)Document23 pagesOrganic Pharmaceutical Chemistry IV 1st Semester, Year 5 (2016-2017)Mohammed AbdullahNo ratings yet
- Pharmaceutical PowdersDocument12 pagesPharmaceutical PowdersThomas Niccolo Filamor ReyesNo ratings yet
- PTT 202 Organic Chemistry Immunological MethodsDocument25 pagesPTT 202 Organic Chemistry Immunological MethodsTanChiaZhiNo ratings yet
- Receptors As Drug Targets PDFDocument66 pagesReceptors As Drug Targets PDFAubreyNo ratings yet
- Bioprocess Engg Lab Assignment: Olivia Newton 119BT0433Document5 pagesBioprocess Engg Lab Assignment: Olivia Newton 119BT0433olivia6669No ratings yet
- Assignment - Screening of Amylase Producing Organisms: Olivia Newton 119BT0433Document3 pagesAssignment - Screening of Amylase Producing Organisms: Olivia Newton 119BT0433olivia6669No ratings yet
- Ethanol Microbial StrainDocument1 pageEthanol Microbial Strainolivia6669No ratings yet
- Assignment - Amylase Production: Olivia Newton 119BT0433Document1 pageAssignment - Amylase Production: Olivia Newton 119BT0433olivia6669No ratings yet
- BPE AssignmentDocument3 pagesBPE Assignmentolivia6669No ratings yet
- Hawaii TravelogueDocument3 pagesHawaii Travelogueolivia6669No ratings yet
- Conversion Schemes in Organic Chemistry: SCHEME - I: Conversions Related To Alkyl HalidesDocument11 pagesConversion Schemes in Organic Chemistry: SCHEME - I: Conversions Related To Alkyl Halidesshephalid_1No ratings yet
- Bio Lab InvestigatoyDocument17 pagesBio Lab Investigatoyolivia6669No ratings yet
- SAT Chem 16e CH 3 Summary PDFDocument1 pageSAT Chem 16e CH 3 Summary PDFolivia6669No ratings yet
- Blah Blah WTF Am I DoingDocument1 pageBlah Blah WTF Am I Doingolivia6669No ratings yet
- Pharmceutical Chemistry Part 1 (Prelim)Document11 pagesPharmceutical Chemistry Part 1 (Prelim)Majeddah Aliudin TalambunganNo ratings yet
- Temperature Programmed Desorption TPDDocument18 pagesTemperature Programmed Desorption TPDyiyiNo ratings yet
- Understanding Lithium Resources and Recycling for BatteriesDocument3 pagesUnderstanding Lithium Resources and Recycling for BatteriesAlejandra RojasNo ratings yet
- Mark Scheme (Results) January 2022Document25 pagesMark Scheme (Results) January 2022윤소리No ratings yet
- Petrochemicals Flowchart (ICIS)Document1 pagePetrochemicals Flowchart (ICIS)Guido BerdinaNo ratings yet
- Improve Landing Gear Corrosion ProtectionDocument26 pagesImprove Landing Gear Corrosion ProtectionImpress Production PlanningNo ratings yet
- Effect of Sodium Carbonate On Forming Capacity of A SoapDocument6 pagesEffect of Sodium Carbonate On Forming Capacity of A SoapThiagarajan BaluNo ratings yet
- Clay Drain Tile and Perforated Clay Drain Tile: Standard Specification ForDocument6 pagesClay Drain Tile and Perforated Clay Drain Tile: Standard Specification FormatiullahNo ratings yet
- TRW 23925036 02Document9 pagesTRW 23925036 02Eddy GuerreroNo ratings yet
- Instrumental Methods of Analysis: Practical Lab ManualDocument73 pagesInstrumental Methods of Analysis: Practical Lab ManualPriya Bardhan RayNo ratings yet
- 07 Alkaline Phosphatase & Acid PhosphataseDocument12 pages07 Alkaline Phosphatase & Acid PhosphataseFrances FranciscoNo ratings yet
- Ionic and Covalent Compound - 121125Document12 pagesIonic and Covalent Compound - 121125Xandria SabrosoNo ratings yet
- AX400 - Specification V1.2 2020.2.1Document6 pagesAX400 - Specification V1.2 2020.2.1AndresNo ratings yet
- Volumetric Analysis LabDocument2 pagesVolumetric Analysis LabOkera JamesNo ratings yet
- Failure Analysis of Conveyor Pulley ShaftDocument12 pagesFailure Analysis of Conveyor Pulley ShafttuanNo ratings yet
- 2007 HKAL/HKASL Chemistry Examination: Pau Chiu Wah 19.3.2004Document18 pages2007 HKAL/HKASL Chemistry Examination: Pau Chiu Wah 19.3.2004ChemistixNo ratings yet
- Cycle - I Experiment-2 Observation and Calculations Titration I - Standardization of EDTADocument5 pagesCycle - I Experiment-2 Observation and Calculations Titration I - Standardization of EDTAadfg100% (1)
- CBSE Class XII Practicals - Titration of Mohr's SaltDocument5 pagesCBSE Class XII Practicals - Titration of Mohr's SaltAsishNo ratings yet
- Inactivates .: Insulin, Secreted by The Pancreas When Blood Glucose Is High, Stimulates PP-1, WhichDocument5 pagesInactivates .: Insulin, Secreted by The Pancreas When Blood Glucose Is High, Stimulates PP-1, Whichratih puspasariNo ratings yet
- US20080287533A1Document22 pagesUS20080287533A1Maikel Perez NavarroNo ratings yet
- Science 9 - Q2 - Mod3 - IONS-HOW-ARE-THEY-FORMED - VerFinalDocument23 pagesScience 9 - Q2 - Mod3 - IONS-HOW-ARE-THEY-FORMED - VerFinalAbel Emmanuel Solitario CabralesNo ratings yet
- DNA EXTRACTION, (Melvin Pagaran, Markus Quinsay, Elijah Dulatas, Gabriel Mangcoy)Document4 pagesDNA EXTRACTION, (Melvin Pagaran, Markus Quinsay, Elijah Dulatas, Gabriel Mangcoy)John Gabriel MangcoyNo ratings yet
- Straight Objective Type: Part-IDocument4 pagesStraight Objective Type: Part-Iaditya aryaNo ratings yet
- 3RD Term S2 Chemistry-1Document35 pages3RD Term S2 Chemistry-1Rikon Uchiha0% (1)