You might also like
- Patrick4e Ch21 PKDocument46 pagesPatrick4e Ch21 PKnosaybaNo ratings yet
- CH 21Document48 pagesCH 21maimaiNo ratings yet
- 2006-CHM6108 - L5L6 SlidesDocument52 pages2006-CHM6108 - L5L6 Slidesaidar.seralinNo ratings yet
- Metabolic Pathways: Glycolysis Pentose Phosphate PathwayDocument1 pageMetabolic Pathways: Glycolysis Pentose Phosphate PathwayLucasLeãoNascimentoNo ratings yet
- 2 Target Obat - 2022Document95 pages2 Target Obat - 2022Yeremi EgaNo ratings yet
- Osteoprorosis Therapy Management, Beyond Safety and Efficacy (Once Yearly Treatment)Document29 pagesOsteoprorosis Therapy Management, Beyond Safety and Efficacy (Once Yearly Treatment)ian ismeNo ratings yet
- 10 Nucleic Acids and Proteins SynthesisDocument60 pages10 Nucleic Acids and Proteins Synthesisjaya1129No ratings yet
- Biosynthesis of Fatty Acids #1Document68 pagesBiosynthesis of Fatty Acids #1QOTRUNNADA AIDANo ratings yet
- Regulatory Enzyme (Enzymatic Regulation) : (WEEK 6)Document15 pagesRegulatory Enzyme (Enzymatic Regulation) : (WEEK 6)oczhinviaNo ratings yet
- What Is Organic Chemistry?Document72 pagesWhat Is Organic Chemistry?gs7514424No ratings yet
- Nucleic Acid Structure & DNA ReplicationDocument51 pagesNucleic Acid Structure & DNA ReplicationJMCDUFFIENo ratings yet
- 14glucose6 PpsDocument2 pages14glucose6 PpsSanjeefKumrIINo ratings yet
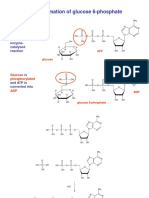
- Formation of Glucose 6-PhosphateDocument2 pagesFormation of Glucose 6-PhosphateSanjeefKumrIINo ratings yet
- Transcription Transcription - Translation Information Flow in Biological Systems - DNA Replication PDFDocument90 pagesTranscription Transcription - Translation Information Flow in Biological Systems - DNA Replication PDFAveen Shaban100% (1)
- Chapter 4 ContiuedDocument24 pagesChapter 4 ContiuedumarNo ratings yet
- Uric Acid Metabolism & GoutDocument44 pagesUric Acid Metabolism & GoutSAIMA PARVEENNo ratings yet
- Lec 5Document20 pagesLec 5Sreemanti DeyNo ratings yet
- Presentazione Tesi Matteo CerrinaDocument82 pagesPresentazione Tesi Matteo CerrinaMatteo CerrinaNo ratings yet
- Energy Production: Hapter 3Document18 pagesEnergy Production: Hapter 3Supriya A SNo ratings yet
- Antibiotics Deraya 2Document33 pagesAntibiotics Deraya 2ahmed montaserNo ratings yet
- The β - D Glucose Scaffold as a β- Turn Mimetic: Saidulu DaraDocument19 pagesThe β - D Glucose Scaffold as a β- Turn Mimetic: Saidulu Daraglreddy09No ratings yet
- 5.36 Biochemistry Laboratory: Mit OpencoursewareDocument11 pages5.36 Biochemistry Laboratory: Mit OpencoursewareNeenu RajputNo ratings yet
- DNA PPSXDocument19 pagesDNA PPSXTintin Brusola SalenNo ratings yet
- Questions: Further ReadingDocument1 pageQuestions: Further ReadingAmir ali WalizadehNo ratings yet
- Nucleotide Metabolism - Part 1 (Purine Biosynthesis)Document49 pagesNucleotide Metabolism - Part 1 (Purine Biosynthesis)Mohammed Ismail HegazyNo ratings yet
- Biosynthetic Pathway of PenicillinsDocument1 pageBiosynthetic Pathway of PenicillinskuganatsukiNo ratings yet
- 2006-CHM6108 - L5L6 HandoutDocument9 pages2006-CHM6108 - L5L6 Handoutaidar.seralinNo ratings yet
- Curs 12 GlycogenDocument37 pagesCurs 12 GlycogenStanescuRozicaNo ratings yet
- Purine Metabolism II: Osama YousefDocument14 pagesPurine Metabolism II: Osama YousefWan NurfazliyanaNo ratings yet
- Curs 12 GlycolysisDocument48 pagesCurs 12 GlycolysisTeodora MunteanuNo ratings yet
- Biopolymers LectureDocument23 pagesBiopolymers LectureNneka Okafor-EzeaniNo ratings yet
- Honey Health Benefits and Uses in Medicine: Hana Scepankova, Jorge A. Saraiva, and Letícia M. EstevinhoDocument14 pagesHoney Health Benefits and Uses in Medicine: Hana Scepankova, Jorge A. Saraiva, and Letícia M. EstevinhoAndrés Felipe Zapata MurielNo ratings yet
- Lipids 2Document6 pagesLipids 2cumbredinNo ratings yet
- Classes Winter11 30CID18 LipidsCh26Document21 pagesClasses Winter11 30CID18 LipidsCh26Daniel TranNo ratings yet
- Kami Export - 6 Biological MoleculesDocument5 pagesKami Export - 6 Biological MoleculesNicholas Crowell100% (1)
- Alkaloid S1Document142 pagesAlkaloid S1Faisal MNo ratings yet
- 9 Glycogen 1Document37 pages9 Glycogen 1micklemagdy50No ratings yet
- DNA ReplicationDocument20 pagesDNA ReplicationFrie An PanteNo ratings yet
- Nucleic Acids: (Deoxyribonucleic Acids) (Ribonucleic Acids)Document45 pagesNucleic Acids: (Deoxyribonucleic Acids) (Ribonucleic Acids)Darshan TrivediNo ratings yet
- Chemistry NoteDocument27 pagesChemistry NoteUmar M abubakarNo ratings yet
- K7 - Metabolisme NukleotidaDocument33 pagesK7 - Metabolisme NukleotidaAditya MuchayatsyahNo ratings yet
- K7 - Metabolisme NukleotidaDocument32 pagesK7 - Metabolisme NukleotidaTiara YosephaNo ratings yet
- Prodrug (D.ashowq)Document4 pagesProdrug (D.ashowq)علي الطياريNo ratings yet
- Nucleotides and Nucleic Acids: CH HO Base P O O O O CH Base P O O O O CH BaseDocument12 pagesNucleotides and Nucleic Acids: CH HO Base P O O O O CH Base P O O O O CH Basebiagio castronovoNo ratings yet
- Lec14 PDFDocument11 pagesLec14 PDFAishwarya MankaniNo ratings yet
- (2022 11 17) 張訓碩-生藥學 (terpenoids)Document64 pages(2022 11 17) 張訓碩-生藥學 (terpenoids)tw910124No ratings yet
- 02 - Haze FormationDocument22 pages02 - Haze FormationGiorgi GhambashidzeNo ratings yet
- 6.0 Nucleotide Metabolism 2019Document60 pages6.0 Nucleotide Metabolism 2019Emmanuel ChendaNo ratings yet
- Molecular Facts and FiguresDocument9 pagesMolecular Facts and FiguresLuc JeronNo ratings yet
- Tetrodotoxin: TTX: BackgroundDocument12 pagesTetrodotoxin: TTX: BackgroundMarrauNo ratings yet
- Bioorganic Chemistry and Biochemistry CHM3218 Summer C 2008: Class WebsiteDocument45 pagesBioorganic Chemistry and Biochemistry CHM3218 Summer C 2008: Class WebsiteMadhu MattaNo ratings yet
- Answers ch10Document6 pagesAnswers ch10김아진No ratings yet
- Available Compound As A Starting Material: Oxalic Acid Glyxol As Aqueous Solution Amino Acid Co2HDocument10 pagesAvailable Compound As A Starting Material: Oxalic Acid Glyxol As Aqueous Solution Amino Acid Co2HAfifaNo ratings yet
- Fatty Acid Oxidation: Molecular Biochemistry IIDocument39 pagesFatty Acid Oxidation: Molecular Biochemistry IIDozdi100% (1)
- Glycogen Synthesis 2Document39 pagesGlycogen Synthesis 2CLEMENTNo ratings yet
- 10 - Beer Stabilization Technology-Clearly A Matter of ChoiceDocument42 pages10 - Beer Stabilization Technology-Clearly A Matter of ChoiceGiorgi GhambashidzeNo ratings yet
- Biological MacromoleculesDocument8 pagesBiological MacromoleculesRintuNo ratings yet
- 025b MetabolismDocument26 pages025b MetabolismSylvia AngelinaNo ratings yet
- 9 GlycogenDocument36 pages9 GlycogenSneha Sagar SharmaNo ratings yet
- Medicinal Chemistry: Lectures Note 10Document20 pagesMedicinal Chemistry: Lectures Note 10nosaybaNo ratings yet
- Medicinal Chemistry: Lectures Note 8Document10 pagesMedicinal Chemistry: Lectures Note 8nosaybaNo ratings yet
- Medicinal Chemistry: Lectures Note 3Document7 pagesMedicinal Chemistry: Lectures Note 3nosaybaNo ratings yet
- Medicinal Chemistry: Lectures Note 4Document22 pagesMedicinal Chemistry: Lectures Note 4nosaybaNo ratings yet
- Lecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12Document28 pagesLecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12nosaybaNo ratings yet
- Lecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12Document28 pagesLecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12nosaybaNo ratings yet
- Lecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12Document28 pagesLecture Note 5: Patrick: An Introduction To Medicinal Chemistry - ch12nosaybaNo ratings yet
- Drug Study - CefpodoximeDocument4 pagesDrug Study - CefpodoximeIzza DeloriaNo ratings yet
- DobutamineDocument2 pagesDobutamineJaessa FelicianoNo ratings yet
- CempakaDocument3 pagesCempakaMangos MudaNo ratings yet
- PH CM 1 Cu 2 - Pharmacodynamics - 1Document10 pagesPH CM 1 Cu 2 - Pharmacodynamics - 1Chesca DomingoNo ratings yet
- ChloroquineDocument17 pagesChloroquineMarcNo ratings yet
- Drugs of AbuseDocument3 pagesDrugs of AbuseNovutry SiregarNo ratings yet
- NUR 207 Drug Dosage Calculation Practice Fall 2020Document5 pagesNUR 207 Drug Dosage Calculation Practice Fall 2020Francesca LopesNo ratings yet
- Guidance On The Rational Use of RH Kit 3 (Post Rape Management)Document8 pagesGuidance On The Rational Use of RH Kit 3 (Post Rape Management)hiba.khellowNo ratings yet
- MODULE 2 - Nursing Process in PharmacologyDocument5 pagesMODULE 2 - Nursing Process in PharmacologyDonna MarieNo ratings yet
- Persistent Depressive Disorder (Dysthymia) - ClinicalKeyDocument13 pagesPersistent Depressive Disorder (Dysthymia) - ClinicalKeyMasithaNo ratings yet
- The Role of Medications in Weight LossDocument14 pagesThe Role of Medications in Weight LossWareesa JamshedNo ratings yet
- 1 +briantonoDocument11 pages1 +briantonoherlin arlikaNo ratings yet
- Unit Ii. Drug Nomenclature, Standards and ClassesDocument11 pagesUnit Ii. Drug Nomenclature, Standards and ClassesValeria GalațanNo ratings yet
- 1 Trigeminal Neuralgia An Overview From Pathophysiology To Pharmacological TreatmentDocument18 pages1 Trigeminal Neuralgia An Overview From Pathophysiology To Pharmacological TreatmentGabriela Elizabeth Gomez CardenasNo ratings yet
- Philippine Pediatric Society, INC. Committee On Research Forums and Workshop (FORM CR-100)Document3 pagesPhilippine Pediatric Society, INC. Committee On Research Forums and Workshop (FORM CR-100)OsMak PediatricsNo ratings yet
- Interaksi Obat AntiaritmiaDocument24 pagesInteraksi Obat AntiaritmiaDian Ulfa Rianda96No ratings yet
- Opthalma Passmedicin 2020Document157 pagesOpthalma Passmedicin 2020VikrantNo ratings yet
- SHC Heme Onc Antimicrobial ProphylaxisDocument4 pagesSHC Heme Onc Antimicrobial ProphylaxisTala MahmoudNo ratings yet
- DR Saleem BotANYDocument15 pagesDR Saleem BotANYsaleemNo ratings yet
- ENGL250 Practice Problems 5Document6 pagesENGL250 Practice Problems 5jam skiNo ratings yet
- English A SBA FormatDocument7 pagesEnglish A SBA FormatYvonnie TzulNo ratings yet
- Protein BindingDocument22 pagesProtein BindingDeepakNo ratings yet
- Pediatric Cardiac Arrest Algorithm: Yes NoDocument1 pagePediatric Cardiac Arrest Algorithm: Yes NoRatna TambaNo ratings yet
- Insight of Nanomedicine Strategies For A Targeted Del 2021 Journal of Drug DDocument16 pagesInsight of Nanomedicine Strategies For A Targeted Del 2021 Journal of Drug DdianaNo ratings yet
- NUCLEAR MEDICINE POLICY 2018 (Edited)Document100 pagesNUCLEAR MEDICINE POLICY 2018 (Edited)Aizat KamalNo ratings yet
- Experiment NoDocument11 pagesExperiment NoKipa AmangNo ratings yet
- HB Psychiatry QuestionsDocument4 pagesHB Psychiatry QuestionsOmesh PrathirajaNo ratings yet
- Basics of Veterinary Compounding: Presented By: Stephanie Montoya, Mba, BS, CPHTDocument38 pagesBasics of Veterinary Compounding: Presented By: Stephanie Montoya, Mba, BS, CPHTMadeline100% (1)
- AdrenalineDocument20 pagesAdrenalinecreatativeNo ratings yet
- Case - ViedyaDocument40 pagesCase - ViedyaRaka WibisonoNo ratings yet