You might also like
- Alien TechnologyDocument11 pagesAlien TechnologySharvari TaklikarNo ratings yet
- Ensuring The Safety of Marketed Medical DevicesDocument77 pagesEnsuring The Safety of Marketed Medical DevicesciocarliaNo ratings yet
- Neill PresentationDocument12 pagesNeill PresentationionadavisNo ratings yet
- Undeniable Reasons People Hate Ivd Test Kit FactoryDocument4 pagesUndeniable Reasons People Hate Ivd Test Kit FactoryplefulppjsNo ratings yet
- FDA Draft Guidance Explains Voluntary Malfunction Summary Reporting ProgramDocument18 pagesFDA Draft Guidance Explains Voluntary Malfunction Summary Reporting ProgramTumma RamaraoNo ratings yet
- Guidance Device MDDSDocument9 pagesGuidance Device MDDSjerushaw.sinapiNo ratings yet
- MHRA Reg 2023Document2 pagesMHRA Reg 2023dandies-slights-0eNo ratings yet
- Combating Counterfeit Drugs: FDA Report on Securing Drug SupplyDocument37 pagesCombating Counterfeit Drugs: FDA Report on Securing Drug SupplynephylymNo ratings yet
- Ykoz 577Document12 pagesYkoz 577YuliaKozlenkoNo ratings yet
- Design Considerations and Pre Market Submission Recommendations For Interoperable Medical Devices - Guidance For Industry and Food and Drug Administration StaffDocument21 pagesDesign Considerations and Pre Market Submission Recommendations For Interoperable Medical Devices - Guidance For Industry and Food and Drug Administration StaffQuality JiveNo ratings yet
- NABIDH Policies & Standards - Clinical Data Coding Terminology Standards20221058155Document16 pagesNABIDH Policies & Standards - Clinical Data Coding Terminology Standards20221058155akaswinrajkNo ratings yet
- Advancing Digital Health FDA Innovation During COVID-19Document3 pagesAdvancing Digital Health FDA Innovation During COVID-19Lucien DedravoNo ratings yet
- Arnold Ventures FDA Draft Guidance DCT Aug 2023Document3 pagesArnold Ventures FDA Draft Guidance DCT Aug 2023Arnold VenturesNo ratings yet
- The Medical Device Regulation ("MDR") - FTI ConsultingDocument6 pagesThe Medical Device Regulation ("MDR") - FTI ConsultingsevgisozugecerNo ratings yet
- SafetyDocument15 pagesSafetyBetty MollaNo ratings yet
- Mobile Health Applications Wpcvd002 0816Document5 pagesMobile Health Applications Wpcvd002 0816Cao Minh TríNo ratings yet
- Facilitating Trade in Vaccines and Essential Medical Supplies: Guidance NoteFrom EverandFacilitating Trade in Vaccines and Essential Medical Supplies: Guidance NoteNo ratings yet
- Innovation Agenda: February 2015Document4 pagesInnovation Agenda: February 2015AdvaMedLCINo ratings yet
- Human Factors Medical Devices v2.0Document35 pagesHuman Factors Medical Devices v2.0EKNo ratings yet
- Breakthrough Devices ProgramDocument31 pagesBreakthrough Devices Programjustlive0630No ratings yet
- USFDA Guidance For Industry - PSUR - What To ReportDocument7 pagesUSFDA Guidance For Industry - PSUR - What To ReportErshad Shafi AhmedNo ratings yet
- Guideline-Conditional Approval For Covid-19 RTK Self-Test - 120821 - V6Document14 pagesGuideline-Conditional Approval For Covid-19 RTK Self-Test - 120821 - V6James Ching-Hoong LiewNo ratings yet
- Food Control System Assessment Tool: Dimension D – Science/Knowledge Base and Continuous ImprovementFrom EverandFood Control System Assessment Tool: Dimension D – Science/Knowledge Base and Continuous ImprovementNo ratings yet
- FDA's Risk-Based Approach to Human Factors in Medical Device Marketing SubmissionsDocument26 pagesFDA's Risk-Based Approach to Human Factors in Medical Device Marketing SubmissionsTumma RamaraoNo ratings yet
- Relibility in Medical Device IndustryDocument14 pagesRelibility in Medical Device IndustryFrancisco Bernal GómezNo ratings yet
- 2015 Innovation AgendaDocument1 page2015 Innovation AgendaAdvaMedLCINo ratings yet
- FDA Communications Oversight in A Digital EraDocument20 pagesFDA Communications Oversight in A Digital EraFreedom MonkNo ratings yet
- Cost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesFrom EverandCost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesNo ratings yet
- Prescription Medication Biopharmaceuticals Blood Transfusions Electromagnetic Radiation Public Health Service ActDocument13 pagesPrescription Medication Biopharmaceuticals Blood Transfusions Electromagnetic Radiation Public Health Service Actmahendra gunjalNo ratings yet
- Mandatory Problem ReportingDocument24 pagesMandatory Problem ReportingTZ LABNo ratings yet
- Medical Device Quality Systems Manual: A Small Entity Compliance GuideDocument468 pagesMedical Device Quality Systems Manual: A Small Entity Compliance GuideScott BeachNo ratings yet
- Medical Device Data Systems, Medical Image Storage Device, and Medical Image Communications Devices (MDDS)Document9 pagesMedical Device Data Systems, Medical Image Storage Device, and Medical Image Communications Devices (MDDS)Qi LiuNo ratings yet
- FDA Oversight of Medical DevicesDocument53 pagesFDA Oversight of Medical Devicesaysh2383100% (1)
- 2018 and Beyond Outlook and Turning PointsDocument40 pages2018 and Beyond Outlook and Turning PointsraytjanNo ratings yet
- NEWS CENTER Maine (NCM) Sent A List of Questions To The FDA and These Were Their ResponsesDocument2 pagesNEWS CENTER Maine (NCM) Sent A List of Questions To The FDA and These Were Their ResponsesNEWS CENTER MaineNo ratings yet
- Healthcare Client Alert KUWAITDocument4 pagesHealthcare Client Alert KUWAITRipunjoy GoswamiNo ratings yet
- Guidance: Updated Letter To Industry On COVID-19Document2 pagesGuidance: Updated Letter To Industry On COVID-19Rand OmNo ratings yet
- Innovation in HealthcareDocument21 pagesInnovation in HealthcareFatima Khalid100% (1)
- PharmStars Announces Spring 2022 Accelerator Graduates: 11 Digital Health Startups Have Successfully Completed PharmStars' Pharma-Focused Accelerator ProgramDocument4 pagesPharmStars Announces Spring 2022 Accelerator Graduates: 11 Digital Health Startups Have Successfully Completed PharmStars' Pharma-Focused Accelerator ProgramPR.comNo ratings yet
- 22013-Retail-Rm3 Covid 19 Guidance NoticeDocument10 pages22013-Retail-Rm3 Covid 19 Guidance NoticearyanNo ratings yet
- Assessment Framework for DigitalToolsDocument25 pagesAssessment Framework for DigitalToolssandeepNo ratings yet
- In Focus 2022: Data, Analytics and Delivery For ImpactDocument44 pagesIn Focus 2022: Data, Analytics and Delivery For ImpactJustus AshabaNo ratings yet
- Medical Device Quality Systems Manual: A Small Entity Compliance GuideDocument7 pagesMedical Device Quality Systems Manual: A Small Entity Compliance Guidetrungthanhnguyen_83No ratings yet
- Reliability in Medical Device IndustryDocument14 pagesReliability in Medical Device IndustrySubramani KarurNo ratings yet
- GN 07 r2 2 Guidance On Complaint Handling (2022 Nov) - PubDocument8 pagesGN 07 r2 2 Guidance On Complaint Handling (2022 Nov) - PubJEYA KUMARANNo ratings yet
- 34-Standards and Regulatory Considerations (693-714)Document22 pages34-Standards and Regulatory Considerations (693-714)racut_khansatraNo ratings yet
- Patient Safety Thesis PDFDocument6 pagesPatient Safety Thesis PDFasiagroverprovo100% (2)
- Medical Device Trends: Increased Regulation Healthcare Provider Consolidation Aging Population Emerging MarketsDocument6 pagesMedical Device Trends: Increased Regulation Healthcare Provider Consolidation Aging Population Emerging MarketsexperioNo ratings yet
- Supply overview- Medical DevicesDocument2 pagesSupply overview- Medical DevicesMark MercuryNo ratings yet
- Excellence in Diagnostic CareDocument20 pagesExcellence in Diagnostic CareDominic LiangNo ratings yet
- Future Proofing Your Digital Health Regulatory StrategyDocument10 pagesFuture Proofing Your Digital Health Regulatory StrategyAjay GangakhedkarNo ratings yet
- Pharmacovigilance FinalDocument32 pagesPharmacovigilance Finalkuppai396No ratings yet
- Guidance Clinical Decision SoftwareDocument26 pagesGuidance Clinical Decision Softwarejerushaw.sinapiNo ratings yet
- CDRH2011111 CompanionDx Final Guidance 7-24-14 PDFDocument13 pagesCDRH2011111 CompanionDx Final Guidance 7-24-14 PDFstalker1841No ratings yet
- Thesis Electronic Medical RecordsDocument6 pagesThesis Electronic Medical Recordscarmenmartinezmcallen100% (1)
- Classification of Medical Device Risk Assessment HCDocument28 pagesClassification of Medical Device Risk Assessment HCmichael DariasNo ratings yet
- Overview of ReportsDocument4 pagesOverview of ReportsMuhammad UmerNo ratings yet
- The Fundamentals of CTD & ECTDDocument45 pagesThe Fundamentals of CTD & ECTDRenuNo ratings yet
- WHO CSDT SampleDocument120 pagesWHO CSDT SampleYen-Yee Lim JacqNo ratings yet
- Essay TopicsDocument1 pageEssay TopicsRenuNo ratings yet
- Work Update: EU IVDR 2017/746 Learnings: Presented by - Renuka Murmu QA/RA ExecutiveDocument5 pagesWork Update: EU IVDR 2017/746 Learnings: Presented by - Renuka Murmu QA/RA ExecutiveRenuNo ratings yet
- 2 12 1-Mir enDocument1 page2 12 1-Mir enRenuNo ratings yet
- Medical Device SectorDocument52 pagesMedical Device SectorRenu100% (1)
- S7 ManualDocument308 pagesS7 ManualBenjamin MurphyNo ratings yet
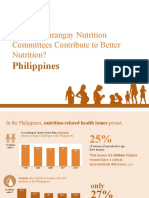
- How Do Barangay Nutrition Committees Contribute To Better Nutrition?Document20 pagesHow Do Barangay Nutrition Committees Contribute To Better Nutrition?Angelito CortunaNo ratings yet
- Depression Thesis StatementDocument4 pagesDepression Thesis Statementsarahmichalakwarren100% (2)
- Potensi Bahaya Fisika, Kimia, Biologi, Ergonomi, Dan Psikologi Pada Tenaga Kerja Di Area Produksi Pabrik GulaDocument5 pagesPotensi Bahaya Fisika, Kimia, Biologi, Ergonomi, Dan Psikologi Pada Tenaga Kerja Di Area Produksi Pabrik GulailhamNo ratings yet
- 12 Rights of Safe Drug AdministrationDocument2 pages12 Rights of Safe Drug Administrationlilcheeza77% (74)
- Prolonged Fasting - How Is It PosiblerDocument17 pagesProlonged Fasting - How Is It PosiblersaturninojonesNo ratings yet
- Carlton Turner StatementDocument1 pageCarlton Turner StatementNews-PressNo ratings yet
- HUMAN REPRODUCTION-1 Madhu - QuestionDocument69 pagesHUMAN REPRODUCTION-1 Madhu - QuestionAyan Sarkar100% (1)
- GE Healthcare India marketing plan for low-resource customersDocument3 pagesGE Healthcare India marketing plan for low-resource customerscharlie tomlinsonNo ratings yet
- Laporan Pendahuluan Polycystic Ovarian Syndrome (PCOS)Document14 pagesLaporan Pendahuluan Polycystic Ovarian Syndrome (PCOS)Jali24No ratings yet
- Clinical and Dermoscopic Features of Seborrheic KeratosisDocument1 pageClinical and Dermoscopic Features of Seborrheic KeratosisRicky SetiawanNo ratings yet
- EpidemiologyDocument100 pagesEpidemiologyKailash NagarNo ratings yet
- Mindfulness and Mental HealthDocument4 pagesMindfulness and Mental Healthkelvin waweruNo ratings yet
- Veterinarians 03-2023Document13 pagesVeterinarians 03-2023PRC BaguioNo ratings yet
- Cure Disease: Charred Skeever Hide Felsaad Tern Feathers Hawk Feathers Mudcrab Chitin Vampire DustDocument5 pagesCure Disease: Charred Skeever Hide Felsaad Tern Feathers Hawk Feathers Mudcrab Chitin Vampire DustFarsiko Risma YandiNo ratings yet
- 20190923swimming Class ProposalDocument9 pages20190923swimming Class ProposalFit AS50% (2)
- FMU 101 - Chap 4 - Test Taking Strategies Part IIDocument11 pagesFMU 101 - Chap 4 - Test Taking Strategies Part IIMario LaraNo ratings yet
- Effects of Disasters and Their ImpactsDocument16 pagesEffects of Disasters and Their ImpactsKrystin DiamosNo ratings yet
- Writing Sample 2Document9 pagesWriting Sample 2api-582848179No ratings yet
- All India Institute of Medical SciencesDocument3 pagesAll India Institute of Medical SciencesNaresh BeatsNo ratings yet
- NCP Self EsteemDocument3 pagesNCP Self EsteemAlfadz AsakilNo ratings yet
- Jeenspsu 17Document13 pagesJeenspsu 17GokulVijayGopal G10No ratings yet
- Principles of Community PsychologyDocument84 pagesPrinciples of Community PsychologyAditya Putra KurniawanNo ratings yet
- Products Category Pack Size MRP (Inr) : Qnet India Product Price List EFFECTIVE January 1st 2020Document2 pagesProducts Category Pack Size MRP (Inr) : Qnet India Product Price List EFFECTIVE January 1st 2020Gopal SainiNo ratings yet
- Home Exercise Program MixedDocument11 pagesHome Exercise Program MixedMadalsa SomayaNo ratings yet
- BSBINN601Document42 pagesBSBINN601Joanne Navarro Almeria78% (23)
- LAP Form v2Document2 pagesLAP Form v2realwavesbhNo ratings yet
- Updated Format SynopsisDocument13 pagesUpdated Format Synopsissiboyif881No ratings yet
- Jean Kazez - The Philosophical Parent - Asking The Hard Questions About Having and Raising Children-Oxford University Press (2017)Document337 pagesJean Kazez - The Philosophical Parent - Asking The Hard Questions About Having and Raising Children-Oxford University Press (2017)Carlos Diaz HuertaNo ratings yet
- COVID Criminal Network Leads To The Gates of HellDocument12 pagesCOVID Criminal Network Leads To The Gates of HellRalf WittkeNo ratings yet