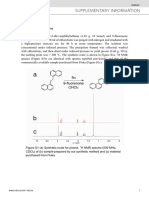
You might also like
- Wis5 WPS 05Document22 pagesWis5 WPS 05Gibson FisherNo ratings yet
- Ombined Footing Design Calculation in Excel SpreadsheetDocument12 pagesOmbined Footing Design Calculation in Excel SpreadsheetfelipeNo ratings yet
- Proton Magnetometer (LG - Huggard) PDFDocument8 pagesProton Magnetometer (LG - Huggard) PDFMario Ariel Vesconi100% (1)
- Exercise Solutions ch.7Document20 pagesExercise Solutions ch.7Tsz Yan Lam86% (7)
- Construction of A Proton Magnetometer: Department of Physics, University of Colombo, Colombo 3Document8 pagesConstruction of A Proton Magnetometer: Department of Physics, University of Colombo, Colombo 3Yunus Emre VarlıNo ratings yet
- RAMAN SPECTROSCOPY Near-InfraredDocument6 pagesRAMAN SPECTROSCOPY Near-InfraredruskaNo ratings yet
- Mathies 1Document8 pagesMathies 1ruskaNo ratings yet
- Mansart Et Al PhysRevB 80 172504 (2009)Document4 pagesMansart Et Al PhysRevB 80 172504 (2009)Daniel SzombatiNo ratings yet
- Tahara 5Document7 pagesTahara 5ruskaNo ratings yet
- Precisely Spun Super Rotors2021nature CommunicationsDocument8 pagesPrecisely Spun Super Rotors2021nature CommunicationsRODRIGO RAMSES DIAZ PACHERRESNo ratings yet
- Tahara 6Document8 pagesTahara 6ruskaNo ratings yet
- Superconductivity, A Physical Chemical Perspective: Ralph C. DoughertyDocument36 pagesSuperconductivity, A Physical Chemical Perspective: Ralph C. Doughertyjuan lopezNo ratings yet
- Nature 07279Document5 pagesNature 07279Marcos San MiguelNo ratings yet
- Zno As A Material Mostly Adapted For The Realization of Room-Temperature Polariton LasersDocument4 pagesZno As A Material Mostly Adapted For The Realization of Room-Temperature Polariton LasersSoufiane BookiNo ratings yet
- Tahara 3Document7 pagesTahara 3ruskaNo ratings yet
- Burda 1999Document6 pagesBurda 1999LAVI TYAGINo ratings yet
- Gupta Spin Coherence in Semiconductor Quantum DotsDocument4 pagesGupta Spin Coherence in Semiconductor Quantum DotsEddy YusufNo ratings yet
- B K Singh Physica Status Solidi A (June 2010)Document6 pagesB K Singh Physica Status Solidi A (June 2010)Tarun YadavNo ratings yet
- Ultrafast Dynamics of Electronic Excitations in A Light-Harvesting Phenylacetylene DendrimerDocument4 pagesUltrafast Dynamics of Electronic Excitations in A Light-Harvesting Phenylacetylene DendrimerGlade680No ratings yet
- CPL222 380Document11 pagesCPL222 380aisman66No ratings yet
- Mathies 3Document10 pagesMathies 3ruskaNo ratings yet
- S.A. Pikuz Et Al - Observation of Laser Satellites in A Plasma Produced by A Femtosecond Laser PulseDocument8 pagesS.A. Pikuz Et Al - Observation of Laser Satellites in A Plasma Produced by A Femtosecond Laser PulsePocxaNo ratings yet
- Interference Measurement of Superposition of Laser-Induced Shock Waves in WaterDocument4 pagesInterference Measurement of Superposition of Laser-Induced Shock Waves in Waterbluefe784294No ratings yet
- High-Pressure Raman Study of Taurine CrystalDocument6 pagesHigh-Pressure Raman Study of Taurine CrystalwpgurgelNo ratings yet
- Observation of Nuclear Fusion Driven by A Pyroelectric CrystalDocument3 pagesObservation of Nuclear Fusion Driven by A Pyroelectric CrystalDavid TurnerNo ratings yet
- Disorder DependentphotoluminescenceinBa0.8Ca0.2TiO3Document6 pagesDisorder DependentphotoluminescenceinBa0.8Ca0.2TiO3Daniel Carvalho de AraújoNo ratings yet
- Cs Na SC F6 CR JLCorrDocument19 pagesCs Na SC F6 CR JLCorrFÍSICA ENSINONo ratings yet
- JChemPhys - 41 - 2403 Thiourea NQRDocument15 pagesJChemPhys - 41 - 2403 Thiourea NQRAllen MNo ratings yet
- Depressed 660-km Discontinuity Caused by Akimotoite-Bridgmanite TransitionDocument12 pagesDepressed 660-km Discontinuity Caused by Akimotoite-Bridgmanite TransitionIgnetious MnisiNo ratings yet
- Vibrational Spectra and Structure Organophosphorous CompoundsDocument8 pagesVibrational Spectra and Structure Organophosphorous CompoundsJack WuNo ratings yet
- Physikalisch-Chemischeslnstitut, Justus-Liebig-Universitiit, Hemrich-Buflring 58, D-6300 Giessen, Federal Republic GermanyDocument3 pagesPhysikalisch-Chemischeslnstitut, Justus-Liebig-Universitiit, Hemrich-Buflring 58, D-6300 Giessen, Federal Republic GermanyRichard Nguedap TegaboueNo ratings yet
- Demonstration of A Nanophotonic Switching Operation by Optical Near-Field Energy TransferDocument4 pagesDemonstration of A Nanophotonic Switching Operation by Optical Near-Field Energy TransferXagreb XaxaNo ratings yet
- Coreno, M 2005 - Branching Ratios in The Radiative Decay of Helium Doubly Excited StatesDocument8 pagesCoreno, M 2005 - Branching Ratios in The Radiative Decay of Helium Doubly Excited StatesAnuvab MandalNo ratings yet
- Introduction To Photochemistry: Ana - Morandeira@fotomol - Uu.seDocument31 pagesIntroduction To Photochemistry: Ana - Morandeira@fotomol - Uu.seSantiago U. NarváezNo ratings yet
- Molecular Shape Effects On Electron and Ion Behavior in Dielectric Liquids: Cis-And Trans-Butene-2 Between The Freezing and Critical PointsDocument7 pagesMolecular Shape Effects On Electron and Ion Behavior in Dielectric Liquids: Cis-And Trans-Butene-2 Between The Freezing and Critical Pointssaurabh_ec21No ratings yet
- DRM12 (2003) 201Document7 pagesDRM12 (2003) 201Gabriel LazarNo ratings yet
- Bulk PCCP 04Document4 pagesBulk PCCP 04felix gütheNo ratings yet
- Phonon Scattering of Excitons and Biexcitons in Zno: K. Hazu and T. SotaDocument3 pagesPhonon Scattering of Excitons and Biexcitons in Zno: K. Hazu and T. SotaEidelsayedNo ratings yet
- High Pressure Raman Study and Phase Transitions of KIO3 Non-Linear Optical Single CrystalsDocument4 pagesHigh Pressure Raman Study and Phase Transitions of KIO3 Non-Linear Optical Single Crystalsarfan.irinbestindoNo ratings yet
- Infrared Spectroscopy: Presented by Uttam Prasad Panigrahy M.PharmDocument40 pagesInfrared Spectroscopy: Presented by Uttam Prasad Panigrahy M.Pharmछेरबहादुर लेउवाNo ratings yet
- 1984 - MORSE - MICHALOPOULOS - Chem - Rev - Spectroscopic Studies of The Jet-Cooled Nickel DimerDocument7 pages1984 - MORSE - MICHALOPOULOS - Chem - Rev - Spectroscopic Studies of The Jet-Cooled Nickel DimerAlejandra AwimbaweNo ratings yet
- Optical Spectroscopy of Tungsten Carbide WC: Shane M. Sickafoose, Adam W. Smith, and Michael D. MorseDocument10 pagesOptical Spectroscopy of Tungsten Carbide WC: Shane M. Sickafoose, Adam W. Smith, and Michael D. MorsessinokrotNo ratings yet
- J Jms 2010 10 003Document8 pagesJ Jms 2010 10 003Rajan PandaNo ratings yet
- AMO Sheet7Document3 pagesAMO Sheet7Ismael CastilloNo ratings yet
- Aiuchi, Shibuya - 2000 - Jet-Cooled Optical Spectroscopy of FeN Between 16 300 and 21 600 CM 1Document27 pagesAiuchi, Shibuya - 2000 - Jet-Cooled Optical Spectroscopy of FeN Between 16 300 and 21 600 CM 1Milan MilovanovićNo ratings yet
- Telfer 2001 0413Document3 pagesTelfer 2001 0413Particle Beam Physics LabNo ratings yet
- All Optical Switching On A Silicon ChipDocument3 pagesAll Optical Switching On A Silicon Chipsimon sanchisNo ratings yet
- Nature08859 s1Document10 pagesNature08859 s1Dejan DjokićNo ratings yet
- Tutorial Chap 9 Ans-CHM580Document2 pagesTutorial Chap 9 Ans-CHM580tirahNo ratings yet
- Inorganic FTIRDocument18 pagesInorganic FTIRAna Paula PereiraNo ratings yet
- Pellegrini 1970 0101Document8 pagesPellegrini 1970 0101Particle Beam Physics LabNo ratings yet
- tmp7EBB TMPDocument3 pagestmp7EBB TMPFrontiers100% (1)
- tmpCF08 TMPDocument4 pagestmpCF08 TMPFrontiersNo ratings yet
- C.I. Frum Et Al - Fourier Transform Emission Spectroscopy of BeF2 at 6.5-Mu-MDocument6 pagesC.I. Frum Et Al - Fourier Transform Emission Spectroscopy of BeF2 at 6.5-Mu-MUasnsdaNo ratings yet
- Search For Time-Reversal Symmetry Breaking in Unconventional SuperconductorsDocument3 pagesSearch For Time-Reversal Symmetry Breaking in Unconventional SuperconductorsrefurlNo ratings yet
- Thermal Conductivity of Yba (Cu ZN) O Single CrystalsDocument7 pagesThermal Conductivity of Yba (Cu ZN) O Single Crystalstestonly261No ratings yet
- S.C. Farantos Et Al - Photofragmentation Spectra of SR + CO Complex: Experiment and Ab Initio CalculationsDocument7 pagesS.C. Farantos Et Al - Photofragmentation Spectra of SR + CO Complex: Experiment and Ab Initio CalculationsMaxnamewNo ratings yet
- 1996 AllOpticalSwitchingLN Stegenab OptLetDocument3 pages1996 AllOpticalSwitchingLN Stegenab OptLetsilviutpNo ratings yet
- Staebler 1980Document8 pagesStaebler 1980Matt OroszNo ratings yet
- Observation of Nonclassical Photon Statistics in The Cavity QED MicrolaserDocument4 pagesObservation of Nonclassical Photon Statistics in The Cavity QED MicrolasercfangyenNo ratings yet
- Long-Distance Single Photon Transmission From A Trapped Ion Via Quantum Frequency ConversionDocument5 pagesLong-Distance Single Photon Transmission From A Trapped Ion Via Quantum Frequency ConversionStephen YinNo ratings yet
- Electric Discharge in Liquids Under The Effect of Vibration: Nikolay, A. BulychevDocument3 pagesElectric Discharge in Liquids Under The Effect of Vibration: Nikolay, A. BulychevDarth FrootLoopsNo ratings yet
- Nondestructive Probing of Rabi Oscillations On The Cesium Clock Transition Near The Standard Quantum LimitDocument4 pagesNondestructive Probing of Rabi Oscillations On The Cesium Clock Transition Near The Standard Quantum LimitWenjun ZhangNo ratings yet
- Tahara 4Document7 pagesTahara 4ruskaNo ratings yet
- Tahara 1Document4 pagesTahara 1ruskaNo ratings yet
- Tahara 3Document7 pagesTahara 3ruskaNo ratings yet
- MicroscopeDocument3 pagesMicroscoperuskaNo ratings yet
- Mathies 3Document10 pagesMathies 3ruskaNo ratings yet
- Mathies 4Document7 pagesMathies 4ruskaNo ratings yet
- Nanotechnology: Kyle Retzer COSC 380Document12 pagesNanotechnology: Kyle Retzer COSC 380ruskaNo ratings yet
- Virtuality of NanotechnologyDocument26 pagesVirtuality of NanotechnologyruskaNo ratings yet
- Raman: It's Not Just For Noodles Anymore: Mario Affatigato Physics Department Coe CollegeDocument48 pagesRaman: It's Not Just For Noodles Anymore: Mario Affatigato Physics Department Coe CollegeruskaNo ratings yet
- Diffractive Optical ElementsDocument8 pagesDiffractive Optical ElementsruskaNo ratings yet
- FSK ScopeDocument4 pagesFSK ScopeHope DlaminiNo ratings yet
- Energies 13 04560Document20 pagesEnergies 13 04560Jay KayeNo ratings yet
- Electrodynamics Introduction: Electrodynamics - Final Exams RevisionDocument27 pagesElectrodynamics Introduction: Electrodynamics - Final Exams RevisionThabelo NgwenyaNo ratings yet
- Calculation Due To High AltitudeDocument1 pageCalculation Due To High AltitudemanishNo ratings yet
- 1.1.1.A.SimpleMachineInvestigation - LeversDocument9 pages1.1.1.A.SimpleMachineInvestigation - LeversSolomon StaplesNo ratings yet
- Steel Lecture - 1Document131 pagesSteel Lecture - 1vinodh159No ratings yet
- Ii (Specti I (& (LLTS) A F.Eading NDT: Traiim (GDocument1 pageIi (Specti I (& (LLTS) A F.Eading NDT: Traiim (Gআশার আলোNo ratings yet
- CHM407 1Document3 pagesCHM407 1Vincent AmobiNo ratings yet
- Second Edition: ISBN: 978-1-4665-0350-2Document452 pagesSecond Edition: ISBN: 978-1-4665-0350-2Fernando FernandezNo ratings yet
- Unit 1 - Home Life N01: Full name: NGUYỄN KHẮC PHÁT Class: 12a1Document9 pagesUnit 1 - Home Life N01: Full name: NGUYỄN KHẮC PHÁT Class: 12a1Phương ThuNo ratings yet
- 6 - Algebraric StructuresDocument43 pages6 - Algebraric StructuresAfiat Khan TahsinNo ratings yet
- Patent Application Publication (10) Pub. No.: US 2009/0126591 A1Document9 pagesPatent Application Publication (10) Pub. No.: US 2009/0126591 A1Quý Đình Mai MaiNo ratings yet
- Toefl Structure ExerciseDocument5 pagesToefl Structure ExerciseNIARAMLINo ratings yet
- Chapter 1 - Mece5104 - Mechanical VibrationDocument22 pagesChapter 1 - Mece5104 - Mechanical VibrationCedricNo ratings yet
- 02-Lignos EtAl-11NCEE ATC PaperRev1Document12 pages02-Lignos EtAl-11NCEE ATC PaperRev1faisaladeNo ratings yet
- StanDocument3 pagesStanVishwaleen RamNo ratings yet
- SGP MCQ Part02Document5 pagesSGP MCQ Part02Rohini HaridasNo ratings yet
- 1.CFD Introduction NotesDocument47 pages1.CFD Introduction Notes刘伟轩No ratings yet
- PDF Examview Waves and Sound Practice Test4 - CompressDocument9 pagesPDF Examview Waves and Sound Practice Test4 - Compressaudrey aurelieNo ratings yet
- Hfu Bachelor Thesis VorlageDocument7 pagesHfu Bachelor Thesis VorlageBuyALiteratureReviewPaperHuntsville100% (2)
- E473Document3 pagesE473sabaris ksNo ratings yet
- All India Test Series For JEE (Advanced) 2024 (For CRP)Document4 pagesAll India Test Series For JEE (Advanced) 2024 (For CRP)Atharva SrivastavaNo ratings yet
- Unit Commitment: Prof. S. Shahnawaz AhmedDocument23 pagesUnit Commitment: Prof. S. Shahnawaz AhmedSaanim MahmoodNo ratings yet
- One Way and Two Way and Cantilever SlabsDocument9 pagesOne Way and Two Way and Cantilever Slabsmariya100% (1)
- Linear and Non-LInear TextDocument2 pagesLinear and Non-LInear TextKorinna PobleteNo ratings yet
- DatasheetDocument13 pagesDatasheetFranki HariminiartoNo ratings yet
- Anbu - Curriculum Vitae PDFDocument5 pagesAnbu - Curriculum Vitae PDFLu SmithNo ratings yet