You might also like
- Development and Validation of A GC-FID Method For The Analysis of Short Chain Fatty Acids in Rat and Human Faeces and in Fermentation FluidsDocument9 pagesDevelopment and Validation of A GC-FID Method For The Analysis of Short Chain Fatty Acids in Rat and Human Faeces and in Fermentation Fluidsjuanda.scienceNo ratings yet
- Dobrowolska-Iwanek Et-Al Hplc-Dad Method For The Quantitative Determination 2020Document7 pagesDobrowolska-Iwanek Et-Al Hplc-Dad Method For The Quantitative Determination 2020Dayanne ZeladaNo ratings yet
- Journal of Chromatography B: SciencedirectDocument9 pagesJournal of Chromatography B: Sciencedirectluis villamarinNo ratings yet
- Journal BiochemistryDocument6 pagesJournal BiochemistrySofiNo ratings yet
- Spirulina (Arthrospira Platensis) Protein-Rich Extract As A Natural Emulsifier For Oil-In-water EmulsionsDocument10 pagesSpirulina (Arthrospira Platensis) Protein-Rich Extract As A Natural Emulsifier For Oil-In-water EmulsionsHerda CahyaningrumNo ratings yet
- Fermentation 04 00079Document9 pagesFermentation 04 00079CHIRANJEEVINo ratings yet
- Cholesterol-Lowering Effects of A Putative Probiotic Strain Lactobacillus Plantarum EM Isolated From Kimchi PDFDocument30 pagesCholesterol-Lowering Effects of A Putative Probiotic Strain Lactobacillus Plantarum EM Isolated From Kimchi PDFpniramolNo ratings yet
- Citoquininas Por HPLCDocument8 pagesCitoquininas Por HPLCeaarizacNo ratings yet
- Phosphotase For Tea - Merged - Compressed (1) - CompressedDocument24 pagesPhosphotase For Tea - Merged - Compressed (1) - Compressedtarun vermaNo ratings yet
- Effect of Nano Silver On GastroprotectivDocument10 pagesEffect of Nano Silver On GastroprotectivSurendra Kumar NekkantiNo ratings yet
- Interactions Between Saccharomyces Cerevisiae and Lactic Acid Bacteria in SourdoughDocument5 pagesInteractions Between Saccharomyces Cerevisiae and Lactic Acid Bacteria in SourdoughmaurodiloretoNo ratings yet
- Food Control: Matheus S. Barbosa, Svetoslav D. Todorov, Cynthia H. Jurkiewicz, Bernadette D.G.M. FrancoDocument7 pagesFood Control: Matheus S. Barbosa, Svetoslav D. Todorov, Cynthia H. Jurkiewicz, Bernadette D.G.M. FrancoLizi SorciaNo ratings yet
- Biotechnology ReportsDocument6 pagesBiotechnology ReportsKevin PaterninaNo ratings yet
- LWT - Food Science and Technology: Guoliang Li, Lihua Dong, Aihong Wang, Wenli Wang, Na Hu, Jinmao YouDocument7 pagesLWT - Food Science and Technology: Guoliang Li, Lihua Dong, Aihong Wang, Wenli Wang, Na Hu, Jinmao YouFRANCIS NDOURNo ratings yet
- Isolation and Identification of Gamma Aminobutyric Acid GA 2013 Current OpiDocument1 pageIsolation and Identification of Gamma Aminobutyric Acid GA 2013 Current OpiEmerald Falah BrayogaNo ratings yet
- 13 - 15v5i2 - 4 Serratia Marcescens OU50TDocument5 pages13 - 15v5i2 - 4 Serratia Marcescens OU50TIsworo RukmiNo ratings yet
- An Effective Solution To Optimise Health Status and Nutrient Utilisation - GUSTOR AQUADocument5 pagesAn Effective Solution To Optimise Health Status and Nutrient Utilisation - GUSTOR AQUAInternational Aquafeed magazineNo ratings yet
- Journal of Chromatography B:, Rikard Landberg, Per Åman, Afaf Kamal-EldinDocument5 pagesJournal of Chromatography B:, Rikard Landberg, Per Åman, Afaf Kamal-EldinNaeem YounisNo ratings yet
- Rapid Screening Procedures For Identification of Succinic Acid ProducersDocument10 pagesRapid Screening Procedures For Identification of Succinic Acid ProducersEdward AlexanderNo ratings yet
- 6610-Texto Do Artigo-25851-1-10-20231229Document9 pages6610-Texto Do Artigo-25851-1-10-20231229moises.martins3No ratings yet
- Lopez Cervantes (Quitina)Document5 pagesLopez Cervantes (Quitina)Sandra MuñozNo ratings yet
- HPLC AcidosDocument5 pagesHPLC AcidosAlejandra Calderón RodríguezNo ratings yet
- Extraction of Bacteriocin and Study of Its Antigonastic AssayDocument6 pagesExtraction of Bacteriocin and Study of Its Antigonastic AssayCrois NadeelNo ratings yet
- BMC BiotechnologyDocument11 pagesBMC Biotechnologyfather45No ratings yet
- Cider Bact PCRDocument6 pagesCider Bact PCRLailatul AzkiyahNo ratings yet
- Characterization and Identification of Lactobacillus Acidophilus Using Biolog Rapid Identification SystemDocument5 pagesCharacterization and Identification of Lactobacillus Acidophilus Using Biolog Rapid Identification Systemarina31No ratings yet
- (MB) Ortiz-Tena2016 Monosaccharides in CVDocument9 pages(MB) Ortiz-Tena2016 Monosaccharides in CVSacra PsyntergiaNo ratings yet
- A Simple, Fast and Sensitive Screening LC-ESI-MSMS Method For Antibiotics in FishDocument9 pagesA Simple, Fast and Sensitive Screening LC-ESI-MSMS Method For Antibiotics in FishyulianaNo ratings yet
- Multi Resi DuosDocument6 pagesMulti Resi DuosNathy GalindoNo ratings yet
- Carbaryl Induced Alterations in Histology and Certain Biochemical Parameters in Liver of Clarias BatrachusDocument5 pagesCarbaryl Induced Alterations in Histology and Certain Biochemical Parameters in Liver of Clarias BatrachusankitNo ratings yet
- Carrageenan Delays Cell Cycle Progression in Human Cancer Cells in Vitro Demonstrated by FUCCI ImaginngDocument9 pagesCarrageenan Delays Cell Cycle Progression in Human Cancer Cells in Vitro Demonstrated by FUCCI ImaginngMarce SiuNo ratings yet
- Salivary Markers of Oxidative Stress and Antioxidant Status: Influence of External FactorsDocument9 pagesSalivary Markers of Oxidative Stress and Antioxidant Status: Influence of External FactorsAndrew NdewNo ratings yet
- LC Determination in Sac Intestine Model PDFDocument12 pagesLC Determination in Sac Intestine Model PDFJose PerezNo ratings yet
- Comparative Real-Time Study of Cellular Uptake of A Formulated Conjugated Linolenic AcidDocument11 pagesComparative Real-Time Study of Cellular Uptake of A Formulated Conjugated Linolenic AcidDebjyoti PaulNo ratings yet
- Isolation and Bioprocess Optimization of HalophiliDocument25 pagesIsolation and Bioprocess Optimization of Halophilisalin MajumdarNo ratings yet
- Aminas Biognicas en Enlatados Lc-MsDocument11 pagesAminas Biognicas en Enlatados Lc-MsKATHERINE LISBETH BERNAL CANALESNo ratings yet
- Cholesterol Assimilation by Lactic Acid Bacteria and Bifidobacteria Isolated From The Human GutDocument5 pagesCholesterol Assimilation by Lactic Acid Bacteria and Bifidobacteria Isolated From The Human GutmicrozasterNo ratings yet
- Food Chemistry: Analytical MethodsDocument7 pagesFood Chemistry: Analytical Methodswildan ariefNo ratings yet
- 10 1016@j LWT 2010 08 011Document8 pages10 1016@j LWT 2010 08 011Tri yatiNo ratings yet
- Pan, X., Chen, F., Wu, T., Tang, H., Dan Zhao, Z. 2009. The Acid, Bile Tolerance and Antimicrobial Property of Lactobacillus Acidophilus NIT. J. Food Control. 20 598-602.Document5 pagesPan, X., Chen, F., Wu, T., Tang, H., Dan Zhao, Z. 2009. The Acid, Bile Tolerance and Antimicrobial Property of Lactobacillus Acidophilus NIT. J. Food Control. 20 598-602.AfdhalRuslanNo ratings yet
- Quantification of Organic Acids by HPLCDocument5 pagesQuantification of Organic Acids by HPLCLee HaronNo ratings yet
- Arce MainDocument9 pagesArce MainDeavid PrietoNo ratings yet
- MPNDocument15 pagesMPNShendi SuryanaNo ratings yet
- Validasi Vit C PDFDocument5 pagesValidasi Vit C PDFFeslyAnugerahAriestaPayungNo ratings yet
- Agrawal Et Al. 2006Document9 pagesAgrawal Et Al. 2006Ivon Buitrago VillanuevaNo ratings yet
- C0 Formulir PemohonanDocument5 pagesC0 Formulir PemohonanDiah AyuningrumNo ratings yet
- Article 1Document6 pagesArticle 1aito7ytNo ratings yet
- Biology 10 00799Document17 pagesBiology 10 00799Pedro GómezNo ratings yet
- 2010 Selection of Local Extremophile Lactic Acid Bacteria WithDocument6 pages2010 Selection of Local Extremophile Lactic Acid Bacteria WithТаро и Астрология с Anatoly KartNo ratings yet
- 1 s2.0 S0023643814000425 MainDocument8 pages1 s2.0 S0023643814000425 MainTHÁI TÂN Nguyễn Hà TrangNo ratings yet
- Articulo 06Document7 pagesArticulo 06iria.gonzalez.micoNo ratings yet
- Chatzimichalakis 2004Document8 pagesChatzimichalakis 2004Pamela Agredo SaninNo ratings yet
- 1 s2.0 S0731708501006112 MainDocument10 pages1 s2.0 S0731708501006112 MainUday BaruahNo ratings yet
- Chemical Implications and Time Reduction of On-Farm Cocoa Fermentation byDocument10 pagesChemical Implications and Time Reduction of On-Farm Cocoa Fermentation byLeonardo Ramos GNo ratings yet
- Assessing Sialic Acid Content in Food by Hydrophilic Chromatography High Performance Liquid ChromatographyDocument7 pagesAssessing Sialic Acid Content in Food by Hydrophilic Chromatography High Performance Liquid ChromatographySze JackNo ratings yet
- Jurnal PenelitianDocument8 pagesJurnal PenelitianmiminNo ratings yet
- Becker 2021Document13 pagesBecker 2021roraysideblancoNo ratings yet
- Becker 2021Document13 pagesBecker 2021roraysideblancoNo ratings yet
- Metabolic Dependent and Independent Ph-Drop Shuts Down Virsr Quorum Sensing in Clostridium PerfringensDocument7 pagesMetabolic Dependent and Independent Ph-Drop Shuts Down Virsr Quorum Sensing in Clostridium Perfringenseti apriyantiNo ratings yet
- Carbohydrate Analysis: High Performance Liquid Chromatography and Capillary ElectrophoresisFrom EverandCarbohydrate Analysis: High Performance Liquid Chromatography and Capillary ElectrophoresisNo ratings yet
- CV Ats 1680229672Document1 pageCV Ats 1680229672Al HafisNo ratings yet
- Dok Baru 2020-03-19 09.58.15Document1 pageDok Baru 2020-03-19 09.58.15Aqidatul Izzah FebrianaNo ratings yet
- Dok Baru 2020-09-25 10.32.16Document1 pageDok Baru 2020-09-25 10.32.16Aqidatul Izzah FebrianaNo ratings yet
- Dok Baru 2020-03-28 10.45.07Document1 pageDok Baru 2020-03-28 10.45.07Aqidatul Izzah FebrianaNo ratings yet
- Perubahan Sifat Mikrobiologi Dan Kimiawi Rusip Selama FermentasiDocument8 pagesPerubahan Sifat Mikrobiologi Dan Kimiawi Rusip Selama FermentasiAqidatul Izzah FebrianaNo ratings yet
- 9716 27929 1 PB PDFDocument10 pages9716 27929 1 PB PDFcici angraini angrainiNo ratings yet
- Hasil MasukptnID Januari 2019 SaintekDocument898 pagesHasil MasukptnID Januari 2019 SaintekAqidatul Izzah FebrianaNo ratings yet
- Bio HomeostasisDocument19 pagesBio HomeostasisRainer WibowoNo ratings yet
- Lec 1 - MCDocument15 pagesLec 1 - MCDivyam JainNo ratings yet
- Chitin and Chitosan As Natural Flocculants For Beer ClarificationDocument6 pagesChitin and Chitosan As Natural Flocculants For Beer ClarificationWILLIAM EDUARDO GOMEZ HERNANDEZNo ratings yet
- Full Download Genetic Analysis An Integrated Approach 3rd Edition Sanders Test BankDocument15 pagesFull Download Genetic Analysis An Integrated Approach 3rd Edition Sanders Test Bankdopemorpheanwlzyv100% (42)
- Tablas HTADocument5 pagesTablas HTAdarwinNo ratings yet
- The Goldschmidt ReactionDocument7 pagesThe Goldschmidt ReactionManojlovic VasoNo ratings yet
- Introduction To Biochemistry 1Document10 pagesIntroduction To Biochemistry 1Manelaine AgnoNo ratings yet
- Che 311 PDFDocument7 pagesChe 311 PDFfamouscNo ratings yet
- Data Sheet-Roofstar - enDocument1 pageData Sheet-Roofstar - enContratistas y Servicios GeneralesNo ratings yet
- Cations and AnionsDocument22 pagesCations and AnionsDoe BlackNo ratings yet
- Thermo Mock SolutionsDocument14 pagesThermo Mock Solutionsmanjeet gajbhiyeNo ratings yet
- Papua LNG Upstream Project: External and Internal Coating and Painting SpecificationDocument65 pagesPapua LNG Upstream Project: External and Internal Coating and Painting SpecificationSangaranNo ratings yet
- Item No. Photo Unit Price (RP) Product Description Material Size (C M) Available Color MinimumDocument20 pagesItem No. Photo Unit Price (RP) Product Description Material Size (C M) Available Color Minimumrobiyanto wandooNo ratings yet
- Basf Masteremaco Application GuideDocument15 pagesBasf Masteremaco Application GuideSolomon AhimbisibweNo ratings yet
- Bio Lab Report (G2) PDFDocument10 pagesBio Lab Report (G2) PDFAina NabihaNo ratings yet
- 1.solid Mechanics-Introduction Normal Stress (CE1201)Document35 pages1.solid Mechanics-Introduction Normal Stress (CE1201)Md. Minhazur Rashid AdnanNo ratings yet
- Two Days Workshop - BrochureDocument1 pageTwo Days Workshop - BrochureShrey VaishnavNo ratings yet
- Thesis On Green Synthesis of NanoparticlesDocument4 pagesThesis On Green Synthesis of NanoparticlesAmanda Moore100% (2)
- Common Ion Sheet: Symbols and Charges of Fixed Charge Mono-Atomic IonsDocument2 pagesCommon Ion Sheet: Symbols and Charges of Fixed Charge Mono-Atomic Ionskarl0% (1)
- 2 (G) 2 (G) 3 (G) 3 (G) 2Document6 pages2 (G) 2 (G) 3 (G) 3 (G) 2shishir kafleNo ratings yet
- Stanbiototal Calcium Liquicolor Procedure No. 0150: Expected ValuesDocument2 pagesStanbiototal Calcium Liquicolor Procedure No. 0150: Expected ValuesKeysi FozNo ratings yet
- US4417079 KurarayDocument16 pagesUS4417079 Kuraray黃英婷No ratings yet
- A Level Chemistry Core Practical 13a Iodine-Propanone ReactionDocument5 pagesA Level Chemistry Core Practical 13a Iodine-Propanone ReactionreeNo ratings yet
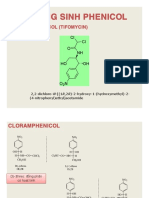
- Khaùng Sinh Phenicol: Cloramphenicol (Tifomycin)Document19 pagesKhaùng Sinh Phenicol: Cloramphenicol (Tifomycin)Dreamline de SkyNo ratings yet
- Hydration Properties of Xylan-Type Structures An FTIR Study of XylooligosaccharidesDocument7 pagesHydration Properties of Xylan-Type Structures An FTIR Study of XylooligosaccharidesvydehiNo ratings yet
- The Structural Components of The Cell Membrane and Its Functions, With Transport Mechanisms.Document17 pagesThe Structural Components of The Cell Membrane and Its Functions, With Transport Mechanisms.Vieyah Angela VicenteNo ratings yet
- Resin Based Restorative Dental Materials. Characteristics and Future PerspectivesDocument13 pagesResin Based Restorative Dental Materials. Characteristics and Future PerspectivesDan MPNo ratings yet
- Production of Biofuel Mini Project PreseDocument18 pagesProduction of Biofuel Mini Project PreseMalike ShamelNo ratings yet
- A1 Management & Technical Consultant Pune: Training Program On Reciprocating CompressorDocument14 pagesA1 Management & Technical Consultant Pune: Training Program On Reciprocating CompressorSandeep KocharNo ratings yet
- Innovations in Semiochemical FormulationDocument20 pagesInnovations in Semiochemical FormulationgfermatuNo ratings yet