You might also like
- Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and GenesFrom EverandNanotechnology-Based Approaches for Targeting and Delivery of Drugs and GenesRating: 5 out of 5 stars5/5 (1)
- London, 22 February 2006 EMEA/CHMP/BMWP/31329/2005: Evaluation of Medicines For Human UseDocument6 pagesLondon, 22 February 2006 EMEA/CHMP/BMWP/31329/2005: Evaluation of Medicines For Human Usealex.pharmathNo ratings yet
- International Conference Harmonisation Technical Requirements Registration Pharmaceuticals Human Use - en 25Document16 pagesInternational Conference Harmonisation Technical Requirements Registration Pharmaceuticals Human Use - en 25Karla GarcíaNo ratings yet
- Guideline Reporting Physiologically Based Pharmacokinetic PBPK Modelling Simulation - enDocument16 pagesGuideline Reporting Physiologically Based Pharmacokinetic PBPK Modelling Simulation - enLay LylyNo ratings yet
- Access New Medicines TR PIO Collaboration ResearchDocument184 pagesAccess New Medicines TR PIO Collaboration ResearchJoana Brazão CachuloNo ratings yet
- 13 ICH S6 Preclinical SafetyDocument10 pages13 ICH S6 Preclinical SafetyKhairulAkmalNo ratings yet
- Guidleine On Potency Testing of Cell Based Immunotherapy Medicinal3Document8 pagesGuidleine On Potency Testing of Cell Based Immunotherapy Medicinal3ИринаNo ratings yet
- London, 18 October 2006 EMEA/CHMP/VWP/164653/2005: European Medicines AgencyDocument19 pagesLondon, 18 October 2006 EMEA/CHMP/VWP/164653/2005: European Medicines AgencyAntonio MoncayoNo ratings yet
- Reflection Paper Methodological Issues Associated Pharmacogenomic Biomarkers Relation Clinical - enDocument21 pagesReflection Paper Methodological Issues Associated Pharmacogenomic Biomarkers Relation Clinical - enmartin.dubuc-extNo ratings yet
- BioTx April 19 2011 SHurstDocument23 pagesBioTx April 19 2011 SHurstSabiruddin Mirza DipuNo ratings yet
- Concept Paper Revision Guideline Clinical Development Vaccines - enDocument3 pagesConcept Paper Revision Guideline Clinical Development Vaccines - enAntonio MoncayoNo ratings yet
- Adopted Reflection Paper Use Extrapolation Development Medicines Paediatrics Revision 1 enDocument20 pagesAdopted Reflection Paper Use Extrapolation Development Medicines Paediatrics Revision 1 en813l1srgr1No ratings yet
- Quality and Equivalence of Topical ProductsDocument5 pagesQuality and Equivalence of Topical Productsblashyrkh_79No ratings yet
- Guideline Similar Biological Medicinal Products Containing Biotechnology Derived Proteins Active - en 2Document13 pagesGuideline Similar Biological Medicinal Products Containing Biotechnology Derived Proteins Active - en 2anjum_niaziNo ratings yet
- Guideline Human Cell Based Medicinal Products - enDocument25 pagesGuideline Human Cell Based Medicinal Products - enИринаNo ratings yet
- Guideline Conduct Bioequivalence Studies Veterinary Medicinal Products Revision 4 - enDocument32 pagesGuideline Conduct Bioequivalence Studies Veterinary Medicinal Products Revision 4 - ensunieljagtap1982No ratings yet
- Analytical Validation of A Novel UHPLC-MS MS Method For 19 Antibiotics Quantification in Plasma Implementation in A LC-MS MS KitDocument14 pagesAnalytical Validation of A Novel UHPLC-MS MS Method For 19 Antibiotics Quantification in Plasma Implementation in A LC-MS MS KitngobaochanNo ratings yet
- S1A GuidelineDocument8 pagesS1A GuidelineJayesh PatilNo ratings yet
- Waters Method DevelopmentDocument50 pagesWaters Method DevelopmentSouradipta GangulyNo ratings yet
- Biopharmaceutics Modelling As A Fundamental Tool To Support Accelerated AccessDocument20 pagesBiopharmaceutics Modelling As A Fundamental Tool To Support Accelerated AccessMaria Rey RicoNo ratings yet
- Ich m9 Biopharmaceutics Classification System Based Biowaivers Step 5 - enDocument18 pagesIch m9 Biopharmaceutics Classification System Based Biowaivers Step 5 - enmagugurgelNo ratings yet
- 5991 1876enDocument32 pages5991 1876enBeatriz ReyesNo ratings yet
- Guideline Quality Aspects Included Product Information Vaccines Human Use Revision 1 - enDocument16 pagesGuideline Quality Aspects Included Product Information Vaccines Human Use Revision 1 - enAbdelkarim BelkebirNo ratings yet
- EDQM PAT Proceedings, 2004Document141 pagesEDQM PAT Proceedings, 2004huynhhaichauchauNo ratings yet
- July 1996 CPMP/ICH/138/95Document9 pagesJuly 1996 CPMP/ICH/138/95MAHESHNo ratings yet
- Docket No. FDA-2016-N-0124: Department of Health and Human ServicesDocument7 pagesDocket No. FDA-2016-N-0124: Department of Health and Human Servicesgerardo sifuentesNo ratings yet
- Miranda2018 PDFDocument38 pagesMiranda2018 PDFDiana LeonNo ratings yet
- Concept Paper Need Revise Guideline Evaluation Anticancer Medicinal Products Man Revision 4 - enDocument3 pagesConcept Paper Need Revise Guideline Evaluation Anticancer Medicinal Products Man Revision 4 - enRob VermeulenNo ratings yet
- Concept Paper Revision Guideline Requirements Clinical Documentation Orally Inhaled Products Oip - enDocument4 pagesConcept Paper Revision Guideline Requirements Clinical Documentation Orally Inhaled Products Oip - enTadilakshmikiranNo ratings yet
- Questions - Answers - Positions On Specific Questionsaddressed Pharmacokinetics Working Party - enDocument48 pagesQuestions - Answers - Positions On Specific Questionsaddressed Pharmacokinetics Working Party - enlhthang1990No ratings yet
- Oecd/Ocde 451: Oecd Guideline For The Testing of ChemicalsDocument15 pagesOecd/Ocde 451: Oecd Guideline For The Testing of ChemicalsVasavi ChittemreddyNo ratings yet
- Oecd/Ocde 451: Oecd Guideline For The Testing of ChemicalsDocument15 pagesOecd/Ocde 451: Oecd Guideline For The Testing of ChemicalsVasavi ChittemreddyNo ratings yet
- Reflection Paper On The Dissolution Specification For Generic Solid Oral Immediate Release Products With Systemic ActionDocument10 pagesReflection Paper On The Dissolution Specification For Generic Solid Oral Immediate Release Products With Systemic ActionAnitha KalyankarNo ratings yet
- AQbD in The BP-Supplementary ChapterDocument16 pagesAQbD in The BP-Supplementary ChapterrbmoureNo ratings yet
- ICH E2E GuidelineDocument20 pagesICH E2E Guidelinetito1628No ratings yet
- Draft Guideline Clinical Investigation Medicinal Products Treatment Prevention Diabetes Mellitus - enDocument24 pagesDraft Guideline Clinical Investigation Medicinal Products Treatment Prevention Diabetes Mellitus - enRafaelNo ratings yet
- Ich-Guidelines of Ich-S. by Samiksha MoreDocument34 pagesIch-Guidelines of Ich-S. by Samiksha MoreSamiksha More100% (1)
- 1.preface The International Pharmacopoeia, Tenth EditionDocument3 pages1.preface The International Pharmacopoeia, Tenth Editionزيد هشام السيدNo ratings yet
- Excipient Risk Assessment: Possible Approaches To Assessing The Risk Associated With Excipient FunctionDocument11 pagesExcipient Risk Assessment: Possible Approaches To Assessing The Risk Associated With Excipient FunctionKolisnykNo ratings yet
- MQA103T LECTURE-3 ICH OverviewDocument19 pagesMQA103T LECTURE-3 ICH OverviewShyam Sunder PancholiNo ratings yet
- From Bioanalytical Method Validation Guidance To ICH M10: Labcorp Drug DevelopmentDocument5 pagesFrom Bioanalytical Method Validation Guidance To ICH M10: Labcorp Drug Developmentlamouna.lamittaNo ratings yet
- Guideline Evaluation Anticancer Medicinal Products Man Revision 5 - enDocument43 pagesGuideline Evaluation Anticancer Medicinal Products Man Revision 5 - enRob VermeulenNo ratings yet
- Guideline On Pharmaceutical Development of Medicines For Paediatric UseDocument23 pagesGuideline On Pharmaceutical Development of Medicines For Paediatric UseJanssen Cariaga ValdezNo ratings yet
- Guideline Repeated Dose Toxicity Revision 1 - enDocument9 pagesGuideline Repeated Dose Toxicity Revision 1 - ennimirani2012No ratings yet
- WHO BreifDocument43 pagesWHO BreifmmssNo ratings yet
- Draft Guideline Safety Efficacy Follow Risk Management Advanced Therapy Medicinal Products Revision - enDocument18 pagesDraft Guideline Safety Efficacy Follow Risk Management Advanced Therapy Medicinal Products Revision - enFDA TaiwanNo ratings yet
- Pedoman OECD No 452 - Toksisitas KronisDocument18 pagesPedoman OECD No 452 - Toksisitas KronisShahifa Audy RahimaNo ratings yet
- Pharm 24 1 1Document2 pagesPharm 24 1 1Darian HerascuNo ratings yet
- Excipient Risk Assessment-Possible Approaches To Assessing The Risk Associated With Excipient FunctionDocument9 pagesExcipient Risk Assessment-Possible Approaches To Assessing The Risk Associated With Excipient FunctionАнна ОрлеоглоNo ratings yet
- Draft Guideline Clinical Evaluation Diagnostic Agents - enDocument16 pagesDraft Guideline Clinical Evaluation Diagnostic Agents - enra bseNo ratings yet
- Guideline Pharmacokinetic Clinical Evaluation Modified Release Dosage Forms - enDocument46 pagesGuideline Pharmacokinetic Clinical Evaluation Modified Release Dosage Forms - enAnton SikorskyiNo ratings yet
- EMA 药品、活性物质、辅料和内包材灭菌指南-2019 (GMP办公室翻译组)Document27 pagesEMA 药品、活性物质、辅料和内包材灭菌指南-2019 (GMP办公室翻译组)于天一No ratings yet
- Combined Chronic Toxicity & Carcinogenicity StudiesDocument20 pagesCombined Chronic Toxicity & Carcinogenicity StudiesLemon DropNo ratings yet
- WC 500090113Document3 pagesWC 500090113Jagdish ChanderNo ratings yet
- Statistical Methods For Clinical Validation of Follow-On Companion Diagnostic Devices Via An External Concordance StudyDocument25 pagesStatistical Methods For Clinical Validation of Follow-On Companion Diagnostic Devices Via An External Concordance StudyWenju QianNo ratings yet
- WC500002770 PDFDocument12 pagesWC500002770 PDFIoana AntonesiNo ratings yet
- Good Practice Guide On Risk Minimisation and Prevention of Medication ErrorsDocument41 pagesGood Practice Guide On Risk Minimisation and Prevention of Medication ErrorsНаталья ИщукNo ratings yet
- Bioanalytical Method Validation - ICHDocument3 pagesBioanalytical Method Validation - ICHfdfsdfdssfsfsNo ratings yet
- Draft Guideline Evaluation Anticancer Medicinal Products Man Revision 5 - enDocument39 pagesDraft Guideline Evaluation Anticancer Medicinal Products Man Revision 5 - enRob VermeulenNo ratings yet
- S1C (R1)Document11 pagesS1C (R1)Anki0391No ratings yet
- Evaluation of Classical, Alternative, and Regulatory Functions of Bone Marrow-Derived MacrophagesDocument11 pagesEvaluation of Classical, Alternative, and Regulatory Functions of Bone Marrow-Derived MacrophagesgugicevdzoceNo ratings yet
- Brain-Eating Ameba (Naegleria)Document2 pagesBrain-Eating Ameba (Naegleria)gugicevdzoceNo ratings yet
- Macrophages in Health and Disease: ReviewDocument21 pagesMacrophages in Health and Disease: ReviewgugicevdzoceNo ratings yet
- Macrophages in Immunoregulation and Therapeutics: Signal Transduction and Targeted TherapyDocument35 pagesMacrophages in Immunoregulation and Therapeutics: Signal Transduction and Targeted TherapygugicevdzoceNo ratings yet
- Urticarial Vasculitis Associated With Essential THDocument3 pagesUrticarial Vasculitis Associated With Essential THgugicevdzoceNo ratings yet
- Mermh1Atotogp.: SbectionDocument1 pageMermh1Atotogp.: SbectiongugicevdzoceNo ratings yet
- The Effects of Atropine and Neostigmine On Heart Rate and RhythmDocument9 pagesThe Effects of Atropine and Neostigmine On Heart Rate and RhythmgugicevdzoceNo ratings yet
- Advances in The Pathogenesis and Treatment of Systemic Lupus ErythematosusDocument23 pagesAdvances in The Pathogenesis and Treatment of Systemic Lupus ErythematosusgugicevdzoceNo ratings yet
- 4-5-6.PATHOMECHANISM - ANGOL 2013aDocument178 pages4-5-6.PATHOMECHANISM - ANGOL 2013azahra eskandarianNo ratings yet
- Current Concepts in The Pathogenesis of Periodontitis: From Symbiosis To DysbiosisDocument20 pagesCurrent Concepts in The Pathogenesis of Periodontitis: From Symbiosis To DysbiosisgugicevdzoceNo ratings yet
- Impact of Systemic Lupus Erythematosus On Oral Health-Related Quality of LifeDocument7 pagesImpact of Systemic Lupus Erythematosus On Oral Health-Related Quality of LifegugicevdzoceNo ratings yet
- Rheumatoid Arthritis: Pathogenesis, Clinical Features, and TreatmentDocument37 pagesRheumatoid Arthritis: Pathogenesis, Clinical Features, and TreatmentgugicevdzoceNo ratings yet
- EeeeDocument2 pagesEeeegugicevdzoceNo ratings yet
- 2Document23 pages2gugicevdzoceNo ratings yet
- Periodontal Disease and Its Association With Systemic DiseaseDocument5 pagesPeriodontal Disease and Its Association With Systemic DiseasegugicevdzoceNo ratings yet
- Xktwu"fgn"fgpiwg "Guvtwevwtc" ("Ekenq"xktcnDocument11 pagesXktwu"fgn"fgpiwg "Guvtwevwtc" ("Ekenq"xktcnFranciscoRamirezNo ratings yet
- Ellis Maitland Jones 1932 Mikulicz S SyndromeDocument1 pageEllis Maitland Jones 1932 Mikulicz S SyndromegugicevdzoceNo ratings yet
- Myxo 1Document8 pagesMyxo 1gugicevdzoceNo ratings yet
- Porter Et Al 1998 Recurrent Aphthous StomatitisDocument16 pagesPorter Et Al 1998 Recurrent Aphthous StomatitisgugicevdzoceNo ratings yet
- Ansible Automation SA Technical Deck Q2FY19Document43 pagesAnsible Automation SA Technical Deck Q2FY19daniel_vp21No ratings yet
- Your Heart: Build Arms Like ThisDocument157 pagesYour Heart: Build Arms Like ThisNightNo ratings yet
- 10 Q - Switching & Mode LockingDocument21 pages10 Q - Switching & Mode Lockingkaushik42080% (1)
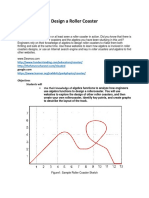
- Design A Roller Coaster ProjectDocument4 pagesDesign A Roller Coaster Projectapi-3564628400% (1)
- Faujifood Pakistan PortfolioDocument21 pagesFaujifood Pakistan PortfolioPradeep AbeynayakeNo ratings yet
- En LF Drivers 10nw76 8Document3 pagesEn LF Drivers 10nw76 8ChrisNo ratings yet
- Guide To Greyhawk PDFDocument108 pagesGuide To Greyhawk PDFAnonymous PtMxUHm9RoNo ratings yet
- Logistics Operation PlanningDocument25 pagesLogistics Operation PlanningLeonard AntoniusNo ratings yet
- Arc 2019-2020Document95 pagesArc 2019-2020AEN HTM DD1 HTM DD1No ratings yet
- Aerodrome Advisory Circular: AD AC 04 of 2017Document6 pagesAerodrome Advisory Circular: AD AC 04 of 2017confirm@No ratings yet
- Laporan Pelayanan Rawat Jalan Tingkat Pertama (RJTP)Document10 pagesLaporan Pelayanan Rawat Jalan Tingkat Pertama (RJTP)dede komalasariNo ratings yet
- Company Profile: Only Milling Since 1967Document16 pagesCompany Profile: Only Milling Since 1967PavelNo ratings yet
- IFIS - Intraoperative Floppy Iris Syndrome Wa Wa 27-09-2008Document18 pagesIFIS - Intraoperative Floppy Iris Syndrome Wa Wa 27-09-2008JanuszNo ratings yet
- Vibrations - NptelDocument3 pagesVibrations - NptelMSK65No ratings yet
- Estimation of Fire Loads For An Educational Building - A Case StudyDocument4 pagesEstimation of Fire Loads For An Educational Building - A Case StudyEditor IJSETNo ratings yet
- Primakuro Catalogue Preview16-MinDocument10 pagesPrimakuro Catalogue Preview16-MinElizabeth LukitoNo ratings yet
- 520L0586 MMF044Document48 pages520L0586 MMF044vendas servicosNo ratings yet
- Diesel Rotary UPS Configurations V1 - 00 - Jan2008Document10 pagesDiesel Rotary UPS Configurations V1 - 00 - Jan2008Karim SenhajiNo ratings yet
- TA1515VDocument4 pagesTA1515VLeo LeiNo ratings yet
- Master of Business Administration in Aviation Management MbaamDocument10 pagesMaster of Business Administration in Aviation Management MbaamAdebayo KehindeNo ratings yet
- Most Important One Liner Questions and Answers May 2022Document15 pagesMost Important One Liner Questions and Answers May 2022pradeepNo ratings yet
- 1 28701-FGC+101+3441+Router+6471+Datasheet+Rev+FDocument2 pages1 28701-FGC+101+3441+Router+6471+Datasheet+Rev+FВладимир ЕгоровNo ratings yet
- Tutorial 2Document2 pagesTutorial 2Adam HakimiNo ratings yet
- Phonetics ReportDocument53 pagesPhonetics ReportR-jhay Mepusa AceNo ratings yet
- CADS Revit Scia Engineer Link Best PracticesDocument32 pagesCADS Revit Scia Engineer Link Best PracticestrevorNo ratings yet
- Total04 Digital Version PDFDocument52 pagesTotal04 Digital Version PDFbeatriz matos67% (3)
- 132kV Substation Feasibility StudyDocument16 pages132kV Substation Feasibility StudyTafadzwa MurwiraNo ratings yet
- Augocom Micro 768 Battery Tester User ManualDocument29 pagesAugocom Micro 768 Battery Tester User ManualJorge PontonNo ratings yet
- XC24M MG DatasheetDocument3 pagesXC24M MG DatasheetAbdulJawad Ibrahim ElmezoghiNo ratings yet