You might also like
- Freud, Sigmund - Cocaine Papers (Stonehill, 1974) PDFDocument459 pagesFreud, Sigmund - Cocaine Papers (Stonehill, 1974) PDFbogdan oșian100% (3)
- Year 2 Drug ListDocument8 pagesYear 2 Drug ListRay100% (1)
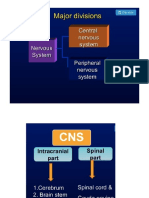
- Drugs Acting On CNSDocument19 pagesDrugs Acting On CNSAditya sagarNo ratings yet
- Neurotransmitter Function Receptor Action Related Drugs/Illnesses Nicotinic Ach-R Curare (N1), Hexamethonium (N2) Atropine (M)Document2 pagesNeurotransmitter Function Receptor Action Related Drugs/Illnesses Nicotinic Ach-R Curare (N1), Hexamethonium (N2) Atropine (M)Yob Ynnos100% (1)
- Sedative-Hypnotics (SeH) and AnxiolyticsDocument101 pagesSedative-Hypnotics (SeH) and Anxiolyticsmatchees-gone rogueNo ratings yet
- (Pharma Tables) Reviewers Compiled PDFDocument115 pages(Pharma Tables) Reviewers Compiled PDFMaverick PascualNo ratings yet
- Veterinary CNS DrugsDocument8 pagesVeterinary CNS DrugsLaureece Salm ApduhanNo ratings yet
- Chapter 30 NotesDocument4 pagesChapter 30 NotesMariah Jane TaladuaNo ratings yet
- The Hitchhiker's Guide To Clinical Pharmacology: Pharmacodynamics: How Drugs WorkDocument23 pagesThe Hitchhiker's Guide To Clinical Pharmacology: Pharmacodynamics: How Drugs WorkMylz MendozaNo ratings yet
- Unit 7. Sympathomimetics and SympatholyticsDocument44 pagesUnit 7. Sympathomimetics and SympatholyticsApril Mergelle Lapuz100% (2)
- Topic 2 - Adrenergic DrugsDocument52 pagesTopic 2 - Adrenergic DrugsAngeli Gregorio100% (1)
- Ectopic PregnancyDocument65 pagesEctopic PregnancyWen SilverNo ratings yet
- Catatonic Schizophrenia NCP (Revised)Document13 pagesCatatonic Schizophrenia NCP (Revised)Wen Silver100% (1)
- PharmacovigilanceDocument31 pagesPharmacovigilanceapi-381097683% (6)
- ValiumDocument1 pageValiumKatie McPeekNo ratings yet
- Sympathomimetic Drugs PharmacologyDocument10 pagesSympathomimetic Drugs PharmacologyHaroon JavedNo ratings yet
- NCM 31112L Midterm Exams: RationalizationDocument202 pagesNCM 31112L Midterm Exams: RationalizationWen SilverNo ratings yet
- Nursing Care of The Older Adult in Chronic IllnessDocument19 pagesNursing Care of The Older Adult in Chronic IllnessWen SilverNo ratings yet
- Pharmacology SummaryDocument14 pagesPharmacology SummaryteddyjolinNo ratings yet
- Adrenergic Agents SeminarDocument48 pagesAdrenergic Agents SeminarTheBoss 20No ratings yet
- 2 Flash CardDocument14 pages2 Flash CardKhem BhattaraiNo ratings yet
- Sympathomimetics and SympatholyticsDocument3 pagesSympathomimetics and Sympatholyticsbunso padillaNo ratings yet
- Copia Di Drugs That Cause or Prolong Delirium - UpToDateDocument2 pagesCopia Di Drugs That Cause or Prolong Delirium - UpToDateAFA.BLSNo ratings yet
- Drugs That May Unmask or Worsen Myasthenia GravisDocument2 pagesDrugs That May Unmask or Worsen Myasthenia GravisnuriajiNo ratings yet
- Sympathomimetic Drugs A. Direct-ActingDocument4 pagesSympathomimetic Drugs A. Direct-ActingZEBINA PIE GENORINGNo ratings yet
- CNS 29 - 39Document12 pagesCNS 29 - 39Michiko_Lois_Tao_203No ratings yet
- Antiparkinsonian DrugsDocument5 pagesAntiparkinsonian Drugsluq9fifNo ratings yet
- Sedative HypnoticDocument60 pagesSedative HypnoticNeeraj KumarNo ratings yet
- 9 - PHAR0004 - 9 - 7 Adrenergics - 1 - RB - 2018 - NotesDocument7 pages9 - PHAR0004 - 9 - 7 Adrenergics - 1 - RB - 2018 - NotesArthi ArumukasamyNo ratings yet
- Pharmacology Classes MindMaps - 230902 - 101709Document73 pagesPharmacology Classes MindMaps - 230902 - 101709mouneeshkumar22No ratings yet
- Practical PharmacologyhjhjhDocument9 pagesPractical PharmacologyhjhjhMonzer AchtarNo ratings yet
- Obat2 Yang Bekerja Pada Ganguan Kesadaran: Elly Usman Bagian Farmakologi Dan Terapi, Fakultas Kedokteran, UnandDocument29 pagesObat2 Yang Bekerja Pada Ganguan Kesadaran: Elly Usman Bagian Farmakologi Dan Terapi, Fakultas Kedokteran, UnandKhairani HakimNo ratings yet
- CNS DrugsDocument8 pagesCNS DrugsLaureece Salm ApduhanNo ratings yet
- Pharmacology Final Drug TablesDocument388 pagesPharmacology Final Drug TablesTJNo ratings yet
- Intro NeurologyDocument64 pagesIntro NeurologySaritha SvNo ratings yet
- Autonomic Nervous SystemDocument54 pagesAutonomic Nervous SystemEzio SartoraNo ratings yet
- 28 - PHAR0004!9!7 Anxiolytic Hypnotic Lecture 2023Document30 pages28 - PHAR0004!9!7 Anxiolytic Hypnotic Lecture 2023Zara KhanNo ratings yet
- 1 - PharmacologyDocument18 pages1 - PharmacologyLuidgi MichelNo ratings yet
- Unit III Drugs Acting On CNSDocument59 pagesUnit III Drugs Acting On CNSZubair khanNo ratings yet
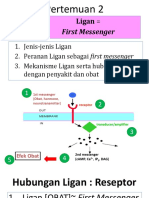
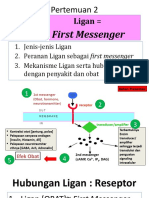
- 2 Ligan First MassengerDocument32 pages2 Ligan First MassengerikhararaNo ratings yet
- Chapter 3Document6 pagesChapter 3SUNKISSED HONEYNo ratings yet
- Adrenergic Agents: University of Negros Occidental-RecoletosDocument66 pagesAdrenergic Agents: University of Negros Occidental-Recoletosmary grace trinidadNo ratings yet
- 2-Ligan-First MassengerDocument33 pages2-Ligan-First MassengerWahid AdvNo ratings yet
- Right-Hand Tremor at Rest, WhichDocument47 pagesRight-Hand Tremor at Rest, WhichsyarintaadeninaNo ratings yet
- Adrenergic Receptor: History CategoriesDocument11 pagesAdrenergic Receptor: History CategoriesUnggul YudhaNo ratings yet
- Margo-7 New - VersionDocument27 pagesMargo-7 New - VersionKhushman KaurNo ratings yet
- 9 - Toxicants That Affect The Autonomic Nervous System (And, in Some Cases, Voluntary NervesDocument71 pages9 - Toxicants That Affect The Autonomic Nervous System (And, in Some Cases, Voluntary NervesCabinet VeterinarNo ratings yet
- CNSDocument102 pagesCNSNoriko MatsumotoNo ratings yet
- Thuốc tđ trên hệ andrenergicDocument3 pagesThuốc tđ trên hệ andrenergicThư PhạmNo ratings yet
- Adrenergic Drugs: Pharmacological Department Medical School - UNPAD Ike HusenDocument33 pagesAdrenergic Drugs: Pharmacological Department Medical School - UNPAD Ike HusenHendra EfendiNo ratings yet
- AdrenomimimeticsDocument40 pagesAdrenomimimeticshealer sruthyNo ratings yet
- College of Nursing: 1 Semester (S.Y. 2019-2020)Document87 pagesCollege of Nursing: 1 Semester (S.Y. 2019-2020)Francis PeterosNo ratings yet
- Sympathomimetic Drug - WikipediaDocument5 pagesSympathomimetic Drug - Wikipediamohammedalradai629No ratings yet
- Drug Class Mechanism of Action Example: ThiazideDocument2 pagesDrug Class Mechanism of Action Example: ThiazideLyod BalagasayNo ratings yet
- Antiemetic Drugs: PHR Sangita ShakyaDocument26 pagesAntiemetic Drugs: PHR Sangita ShakyaCurex QANo ratings yet
- HW 2 Na 3Document7 pagesHW 2 Na 3Araw GabiNo ratings yet
- Antianxiety DrugsDocument56 pagesAntianxiety DrugsVictor AlexandreNo ratings yet
- Sedative-Hypnotics: Pharmacodynamics/ToxicodynamicsDocument12 pagesSedative-Hypnotics: Pharmacodynamics/Toxicodynamicslaras pamekasNo ratings yet
- Antipsychotic AgentsDocument18 pagesAntipsychotic AgentsmengakuNo ratings yet
- Anti-Emetic DrugsDocument10 pagesAnti-Emetic DrugsLydia Alexandra LatumahinaNo ratings yet
- MEDICAL SURGICAL NURSING ASSESSMENT AND MANAGEMENT OF CLINICAL PROBLEMS 9th EditionDocument1 pageMEDICAL SURGICAL NURSING ASSESSMENT AND MANAGEMENT OF CLINICAL PROBLEMS 9th EditionMeryPinkihanNo ratings yet
- Esters of CholineDocument4 pagesEsters of CholineBqunahNo ratings yet
- Pharmacology of Drugs Acting On Sympathetic Nervous SystemDocument65 pagesPharmacology of Drugs Acting On Sympathetic Nervous SystemCharles DapitoNo ratings yet
- 6-Pharma Table 2015Document4 pages6-Pharma Table 2015Fariz AzizNo ratings yet
- Anti-Depressant Drugs: Presented by L.Nithish Shankar Ii Year Mbbs KGMCDocument17 pagesAnti-Depressant Drugs: Presented by L.Nithish Shankar Ii Year Mbbs KGMCÑiťhišh Śhankąŕ LóganáthánNo ratings yet
- 7779087Document65 pages7779087MohamedNo ratings yet
- Case StudyDocument15 pagesCase StudyWen SilverNo ratings yet
- You Are Caring For A Patient With An NG Feeding TubeDocument2 pagesYou Are Caring For A Patient With An NG Feeding TubeWen Silver100% (1)
- AlprazolamDocument10 pagesAlprazolamWen SilverNo ratings yet
- Maladaptive Exam 1Document20 pagesMaladaptive Exam 1Wen SilverNo ratings yet
- Job Satisfaction of Nurses and Their Productivity: Presented by Mba (HM) MD DilnawazDocument22 pagesJob Satisfaction of Nurses and Their Productivity: Presented by Mba (HM) MD DilnawazWen SilverNo ratings yet
- NursingDocument8 pagesNursingWen SilverNo ratings yet
- Drug Study TuberculosisDocument6 pagesDrug Study TuberculosisWen SilverNo ratings yet
- Laboratory and Diagnostic ResultsDocument1 pageLaboratory and Diagnostic ResultsWen SilverNo ratings yet
- Case 1 - Advance DirectivesDocument8 pagesCase 1 - Advance DirectivesWen SilverNo ratings yet
- NCP Risk For Infection TuberculosisDocument3 pagesNCP Risk For Infection TuberculosisWen SilverNo ratings yet
- Health Insurance Portability and Accountability ActDocument14 pagesHealth Insurance Portability and Accountability ActWen SilverNo ratings yet
- The New Food and Drug Administration (Fda) of The Republic of ThephilippinesDocument16 pagesThe New Food and Drug Administration (Fda) of The Republic of ThephilippinesWen SilverNo ratings yet
- Presented By: Agunod, Wengel Banasihan, Samantha Nicole A. Garcia, Lester Maitim, Princess Anne Oarga, Matelyn SDocument24 pagesPresented By: Agunod, Wengel Banasihan, Samantha Nicole A. Garcia, Lester Maitim, Princess Anne Oarga, Matelyn SWen SilverNo ratings yet
- Drug Study WengelDocument3 pagesDrug Study WengelWen SilverNo ratings yet
- The American Psychiatric Publishing Textbook of PsDocument3 pagesThe American Psychiatric Publishing Textbook of Ps!R_I_A!No ratings yet
- Marketing Management Report GSKDocument18 pagesMarketing Management Report GSKavinashkhetpal100% (1)
- Adr Form 10Document3 pagesAdr Form 10doctor uroosaNo ratings yet
- Impact Factor 2021: List of Journals by Clarivate Analytics (JCR)Document264 pagesImpact Factor 2021: List of Journals by Clarivate Analytics (JCR)Medjahed AB100% (1)
- Avigan英文仿單Document6 pagesAvigan英文仿單VenkatNo ratings yet
- Flurazepam Prescribing InformationDocument2 pagesFlurazepam Prescribing InformationMohammed Shamiul ShahidNo ratings yet
- OTC Drugs - Students' Class NotesDocument10 pagesOTC Drugs - Students' Class NotesMonamonaNo ratings yet
- Stock 15 FebDocument15 pagesStock 15 FebnaelarizqiNo ratings yet
- SacubitrilValsartan (Entresto) For Heart FailureDocument2 pagesSacubitrilValsartan (Entresto) For Heart FailureWatchara TansiriNo ratings yet
- Batuk - Pilek Harga Jumlah Barang: Satuan Masuk Terjual SisaDocument7 pagesBatuk - Pilek Harga Jumlah Barang: Satuan Masuk Terjual Sisadoni januarindraNo ratings yet
- No Kode Barang Nama Barang Saldo Awal Beli Retur ResepDocument27 pagesNo Kode Barang Nama Barang Saldo Awal Beli Retur ResepFarmasi Bandung Heart ClinicNo ratings yet
- Biocon CaseDocument2 pagesBiocon CaseDinaraNo ratings yet
- MTAC Assignment GeriatricDocument18 pagesMTAC Assignment Geriatrichanie mansorNo ratings yet
- General ChapterDocument7 pagesGeneral ChapterAhmed Osama ShalashNo ratings yet
- Elixir - WikipediaDocument3 pagesElixir - WikipediaMohamed IbrahimNo ratings yet
- Crash Course Crash Course: Niper-2021Document7 pagesCrash Course Crash Course: Niper-2021ramesh joshiNo ratings yet
- Common Disk-Shaped Tablets A Tablet Is A Mixture of ActiveDocument8 pagesCommon Disk-Shaped Tablets A Tablet Is A Mixture of Activenasser9876No ratings yet
- FIR Quashed Under NDPS Sarhind CaseDocument7 pagesFIR Quashed Under NDPS Sarhind CasesmartpaulNo ratings yet
- Module 8: Preventing Opioid Overdose With Education and Naloxone Rescue KitsDocument23 pagesModule 8: Preventing Opioid Overdose With Education and Naloxone Rescue KitsNarges AlaieNo ratings yet
- Session 1: Introduction To Pharmaceutical Dosage Forms: by NTENGO, Venance Wilfred Bpharm K's Royal CollegeDocument27 pagesSession 1: Introduction To Pharmaceutical Dosage Forms: by NTENGO, Venance Wilfred Bpharm K's Royal CollegeVenance NtengoNo ratings yet
- Price ListDocument15 pagesPrice ListmahinggarNo ratings yet
- Antipsychotic CassificatioonDocument23 pagesAntipsychotic Cassificatioonedelweis_04No ratings yet
- Pricelist Harga ObatDocument22 pagesPricelist Harga ObatNIRMALANo ratings yet
- Medchem 2Document8 pagesMedchem 2Nikhil ThakurNo ratings yet
- Factors InfluenceDocument12 pagesFactors InfluencemayankpdNo ratings yet
- PNS MethoxamineDocument1 pagePNS MethoxamineKim RuizNo ratings yet
- Sanofi PriceListDocument1 pageSanofi PriceListJohn EnochNo ratings yet